一种分级多孔金属有机框架化合物及其制备方法与应用
1.本发明属于分离纯化领域,具体涉及一种分级多孔金属有机框架化合物及其制备方法与应用。
背景技术:
2.甜菊糖苷(sgs)是一种高甜度、低热量的天然甜味剂,提取自甜叶菊叶片,主要用作食品添加剂和医药制剂。sgs中含量最多、甜度最高的成分是莱鲍迪苷a(ra)和甜菊苷(stv),分别约占50
‑
60%和20
‑
30%,其中ra口感甜美,而stv具有不良的后苦味。
3.近年来,为了去除sgs中不良口味的stv获得高纯度的ra,国内外已有很多关于甜菊糖苷分离纯化的研究。目前主要应用的方法有重结晶法,高速逆流色谱法,膜分离法和吸附法。其中吸附法因操作简单,效率高,成本低等优势成为最常用的分离纯化sgs的方法。然而目前的大多数吸附剂为不涉及结构
‑
性能关系的物理/化学吸附。
4.金属有机框架化合物(mof)作为一种由金属离子和有机配体经配位键结合而成的新型多孔材料,具有高比表面积,高孔隙率和易于修饰等特点,可用于天然产物吸附分离。传统的mof大都是微孔结构,只能局限于低分子量小分子物质传输,无法满足stv和ra等分子量在800
‑
1000的小分子物质进入mof孔道。为了解决这一问题,分级多孔mof(hp
‑
mof)的研究和发展拓宽了mof材料的应用领域,hp
‑
mof是指在mof微孔结构基础上原位构建介孔,采用不同的手段以形成不同性质的孔隙。但目前尚未见适宜stv和ra分离的hp
‑
mof材料的相关报道。
技术实现要素:
5.有鉴于此,本发明目的在于提供一种分级多孔金属有机框架化合物hp
‑
nh2‑
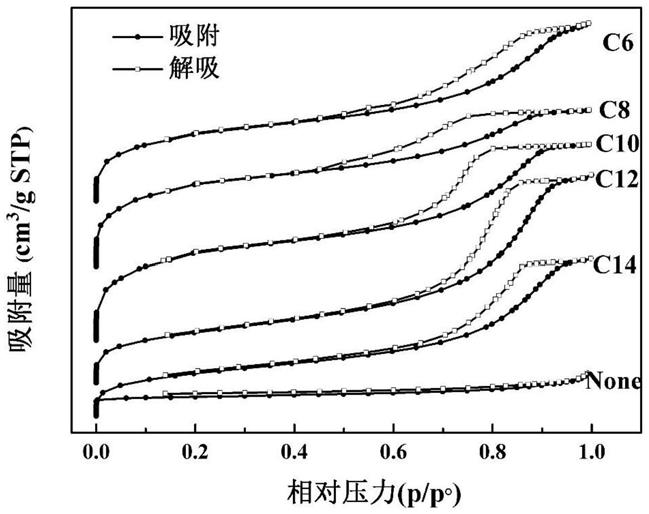
mil
‑
53及其制备方法与应用。本发明通过调节剂诱导缺陷法构建微、介孔的分级多孔结构,实现吸附量和吸附选择性的同时提升,在大量创造性实验的基础上确定适宜甜菊糖苷分离的孔隙结构参数,可用于甜菊糖苷中ra和stv的选择性分离。
6.为达到以上目的,本发明采用如下技术方案:本发明提供了一种分级多孔金属有机框架化合物hp
‑
nh2‑
mil
‑
53,所述的化合物具有可调的缺陷孔,比表面积在175.04~2293.98m2/g之间可调,孔体积在0.45~2.16cm3/g之间可调,孔径在0.38~26.75nm之间可调。
7.本发明还提供了一种分级多孔金属有机框架化合物的制备方法,具体包括以下步骤:(1)称取一定量的alcl3·
6h2o,溶解于适量的溶剂中,加入一定量的调节剂,搅拌加入适量的2
‑
氨基对苯二甲酸,磁力搅拌后倒入聚四氟乙烯内衬的高压反应釜,在一定条件下反应,反应结束后将沉淀物用dmf离心清洗;(2)将步骤(1)中得到的沉淀物在一定条件下活化,之后依次用dmf和甲醇清洗,真空干燥后得到分级多孔金属有机框架化合物hp
‑
nh2‑
mil
‑
53。
8.进一步地,步骤(1)中所述的调节剂包括:己酸,辛酸,癸酸,月桂酸或肉豆蔻酸中的一种。
9.进一步地,步骤(1)中所述的溶剂为dmf;alcl3·
6h2o、溶剂和调节剂的用量比为1mmol:8~20ml:5~15mmol。
10.步骤(1)中所述的alcl3·
6h2o与2
‑
氨基对苯二甲酸的摩尔比为1:0.4~2。
11.步骤(1)中所述的一定条件下反应的反应条件包括:反应温度为120~150℃,反应时长为20~30h。
12.步骤(2)中所述的活化为将沉淀物放入含有一定浓度hcl的dmf溶液中进行活化,所述dmf溶液中hcl与dmf的体积比为1:100~300。
13.步骤(2)中所述活化的条件为60~100℃搅拌8~12h,优选为90℃搅拌10h。
14.步骤(2)中所述真空干燥的温度为150~200℃,时间为12~15h。
15.本发明还提供了上述分级多孔金属有机框架化合物hp
‑
nh2‑
mil
‑
53在甜菊糖苷分离纯化中的应用。
16.优选地,所述应用包括:在锥形瓶中称取一定量的hp
‑
nh2‑
mil
‑
53,加入一定浓度的甜菊糖苷水溶液,在一定条件恒温振荡下吸附甜菊糖苷,采用hplc测定stv和ra的含量。
17.进一步优选地,所述hp
‑
nh2‑
mil
‑
53与甜菊糖苷水溶液的用量比为30~100mg:25~100ml;甜菊糖苷水溶液的浓度为6mg/ml(stv:ra=1:1)。
18.与现有技术相比,本发明的有益效果在于:本发明通过金属离子与过量调节剂首先形成金属
‑
氧簇合物,之后少量低pka的有机配体的加入,可以不完全替换调节剂与金属配位,因此形成可调的缺陷孔。通过改变调节剂的种类和相对含量,系统性探究了孔径分布和比表面积等孔隙参数的变化。本发明制备的hp
‑
nh2‑
mil
‑
53可实现nh2‑
mil
‑
53的比表面积在175.04~2293.98m2/g范围内可调,孔容积在0.45~2.16cm3/g之间可调,和孔径在0.38~26.75nm内可调;筛选出最佳物料比制备的hp
‑
mof
‑
1:10:0.6具有591.31m2/g的比表面积,0.48cm3/g的孔容积和3.80nm的孔径。本发明在不引入特异性基团的情况下,通过孔径筛分作用,选择性地吸附分子量较小的stv,从而实现ra的提纯。进一步地,本发明所提供的化合物具有良好的循环再生能力,加入解吸液恒温振荡可将吸附饱和的hp
‑
nh2‑
mil
‑
53过滤分离出来,降低分离成本。本发明所制备的hp
‑
nh2‑
mil
‑
53不仅为甜菊糖苷分离纯化提供了新材料,还有望为其他相似分子量物质的吸附提供参考,在吸附分离领域具有广阔的应用前景。
附图说明
19.图1为实施例1所制备的hp
‑
nh2‑
mil
‑
53及nh2‑
mil
‑
53的n2吸脱附曲线图;图2为实施例1所制备的hp
‑
nh2‑
mil
‑
53及nh2‑
mil
‑
53的孔径分布图;图3为实施例2所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图;图4为实施例2所制备的hp
‑
nh2‑
mil
‑
53的孔径分布图;图5为实施例3所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图;图6为实施例3所制备的hp
‑
nh2‑
mil
‑
53的孔径分布图;图7为实施例5所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图;图8为实施例5所制备的hp
‑
nh2‑
mil
‑
53的孔径分布图;
图9为实施例6所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图;图10为实施例6所制备的hp
‑
nh2‑
mil
‑
53的孔径分布图。
具体实施方式
20.通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的范围并不限于下述实施例。本发明对试验中所使用到的材料以及试验方法进行一般性和/或具体的描述。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照厂商所建议的条件实施检测。下述实施例中所用的试剂、生物材料等,如无特殊说明,均可从商业途径得到。
21.实施例1本实施例中制备不同调节剂种类的分级多孔金属有机框架化合物,具体制备方法包括如下步骤:(1)称取5份5mmol(1.21g)的alcl3·
6h2o,搅拌溶解于40ml n,n
‑
二甲基甲酰胺(dmf)中,分别加入50mmol的己酸(c6,5.81g)、辛酸(c8,7.21g)、癸酸(c10,8.61g)、月桂酸(c12,10.02g)、肉豆蔻酸(c14,11.42g),搅拌混合后再分别加入5mmol(0.91g)的2
‑
氨基对苯二甲酸(nh2‑
bdc),分别将混合物搅拌30min后倒入聚四氟乙烯内衬的高压反应釜,在130℃的条件下反应24h,反应结束后将各沉淀物用dmf离心清洗三次。
22.(2)分别将步骤(1)中得到的沉淀物置于40ml含有0.2ml hcl的dmf溶液中,在80℃下搅拌12h完成活化过程,用dmf和甲醇依次清洗三次,并在150℃的真空干燥箱中干燥12h,得到不同调节剂种类对应的hp
‑
nh2‑
mil
‑
53。分别记为hp
‑
mof
‑
c6、hp
‑
mof
‑
c8、hp
‑
mof
‑
c10、hp
‑
mof
‑
c12、hp
‑
mof
‑
c14。分别对得到的hp
‑
nh2‑
mil
‑
53进行比表面积测试(bet),bet表征结果表明hp
‑
mof
‑
c6、hp
‑
mof
‑
c8、hp
‑
mof
‑
c10、hp
‑
mof
‑
c12、hp
‑
mof
‑
c14的比表面积分别为:922.14m2/g、1104.39m2/g、1178.14m2/g、706.22m2/g和597.02m2/g;其孔体积分别为:1.05cm3/g、0.92cm3/g、1.15cm3/g、1.22cm3/g和0.93cm3/g;孔径范围分别在0.63
‑
15.67nm、0.64
‑
17.39nm、0.64
‑
21.73nm、1.24
‑
20.20nm和0.63
‑
26.75nm。
23.测试上述制备得到的不同调节剂种类对应的hp
‑
nh2‑
mil
‑
53在30℃条件下的吸附性能。在锥形瓶中各称取30mg的hp
‑
mof
‑
c6、hp
‑
mof
‑
c8、hp
‑
mof
‑
c10、hp
‑
mof
‑
c12、hp
‑
mof
‑
c14,分别加入25ml浓度6mg/ml(stv:ra=1:1)的甜菊糖苷水溶液,在150rpm,30℃条件下恒温振荡24h吸附甜菊糖苷,采用hplc测定stv和ra的含量,各个hp
‑
nh2‑
mil
‑
53取三个平行样品进行测试,所有测试的取值为平行样的加和平均值。经过测试hp
‑
mof
‑
c6、hp
‑
mof
‑
c8、hp
‑
mof
‑
c10、hp
‑
mof
‑
c12、hp
‑
mof
‑
c14对应的stv和ra的总吸附量分别为:166.83mg/g、253.89mg/g、155.32mg/g、106.40mg/g、58.80mg/g。对应的stv相对于ra的吸附选择性分别为:1.34、1.34、1.69、1.81、2.58。
24.作为对比,本实施例中还制备了未添加调节剂的分级多孔金属有机框架化合物(nh2‑
mil
‑
53),其制备步骤与hp
‑
nh2‑
mil
‑
53的区别为不添加调节剂己酸、辛酸、癸酸、月桂酸及肉豆蔻酸,将得到的最终产物记为nh2‑
mil
‑
53。bet结果表明nh2‑
mil
‑
53具有175.04m2/g的比表面积,0.45cm3/g的孔体积和1.09
‑
1.73nm的孔径范围。采用与hp
‑
nh2‑
mil
‑
53相同测试方法测试所得nh2‑
mil
‑
53在30℃条件下的吸附性能。经过测试,其对stv的吸附量为4.20mg/g,对ra没有吸附。图1、图2分别为本实施例所制备的hp
‑
nh2‑
mil
‑
53及nh2‑
mil
‑
53的n2吸脱附曲线图及孔径分布图;由图1、图2可见,调节剂可对缺陷孔的大小产生影响,通过
加入不同种类的调节剂可以制备不同孔径的框架化合物。
25.实施例2本实施例中制备不同调节剂含量的分级多孔金属有机框架化合物,具体制备方法包括如下步骤:(1)称取4份5mmol(1.21g)alcl3·
6h2o,搅拌溶解于40ml的dmf中,每份中分别加入25mmol(4.31g)、40mmol(6.89g)、50mmol(8.61g)、60mmol(10.34g)的癸酸搅拌混合,再分别加入5mmol(0.91g)nh2‑
bdc,将混合物搅拌30min后倒入聚四氟乙烯内衬的高压反应釜,在130℃的条件下反应24h,反应结束后将沉淀物用dmf离心清洗三次。
26.(2)分别将步骤(1)中清洗完的沉淀物置于40ml含有0.2ml hcl的dmf溶液中,在80℃下搅拌12h完成活化过程,最后用dmf和甲醇依次清洗三次,并在150℃的真空干燥箱中干燥12h,得到不同调节剂含量对应的hp
‑
nh2‑
mil
‑
53,分别记为hp
‑
mof
‑
1:5:1、hp
‑
mof
‑
1:8:1、hp
‑
mof
‑
1:10:1(hp
‑
mof
‑
c10)、hp
‑
mof
‑
1:12:1。bet表征结果表明,hp
‑
mof
‑
1:5:1、hp
‑
mof
‑
1:8:1、hp
‑
mof
‑
1:10:1、hp
‑
mof
‑
1:12:1分别具有1998.32m2/g、1900.24m2/g、1178.14m2/g和632.71m2/g的比表面积,1.59cm3/g、2.16cm3/g、1.15cm3/g和0.62cm3/g的孔体积,以及0.64
‑
2.95nm、0.64
‑
3.18nm、1.39
‑
7.69nm和1.39
‑
12.91nm的孔径范围。
27.采用实施例1中的测试方法测试上述制备得到的四种hp
‑
nh2‑
mil
‑
53在30℃条件下的吸附性能,对应hp
‑
mof
‑
1:5:1、hp
‑
mof
‑
1:8:1、hp
‑
mof
‑
1:10:1、hp
‑
mof
‑
1:12:1对stv和ra的总吸附量分别为:161.61mg/g、155.18mg/g、168.39mg/g、159.03mg/g。对应stv相对于ra的吸附选择性分别为:1.49、1.50、1.70、1.63。
28.图3、图4分别为本实施例所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图及孔径分布图。由图3、图4可见,调节剂加入的含量影响缺陷孔的数量,进而影响孔径分布范围。
29.实施例3本实施例中制备不同配体含量的分级多孔金属有机框架化合物,具体制备方法包括如下步骤:(1)称取4份5mmol(1.21g)alcl3·
6h2o,搅拌溶解于40ml的dmf中,每份中加入50mmol(8.61g)癸酸(c10)搅拌混合,再分别加入5mmol(0.91g)、4mmol(0.73g)、3mmol(0.54g)、2mmol(0.36g)的nh2‑
bdc,将混合物搅拌30min后倒入聚四氟乙烯内衬的高压反应釜,在130℃的条件下反应24h,反应结束后将沉淀物用dmf离心清洗三次。
30.(2)分别将步骤(1)中清洗完的沉淀物置于40ml含有0.2ml hcl的dmf溶液中,在80℃下搅拌12h完成活化过程,最后用dmf和甲醇依次清洗三次,并在150℃的真空干燥箱中干燥12h,得到不同配体含量对应的hp
‑
nh2‑
mil
‑
53,分别记为hp
‑
mof
‑
1:10:1、hp
‑
mof
‑
1:10:0.8、hp
‑
mof
‑
1:10:0.6、hp
‑
mof
‑
1:10:0.4。bet表征结果表明hp
‑
mof
‑
1:10:1、hp
‑
mof
‑
1:10:0.8、hp
‑
mof
‑
1:10:0.6、hp
‑
mof
‑
1:10:0.4具有1178.14m2/g、886.60m2/g、591.31m2/g和552.48m2/g的比表面积,1.15cm3/g、1.10cm3/g、0.48cm3/g和0.60cm3/g的孔体积,以及1.48~7.69nm、1.48~7.73nm、1.48~6.46nm和2.63~6.46nm的孔径分布范围,且峰值出现在3.80nm。
31.采用实施例1中的测试方法测试上述制备得到的四种hp
‑
nh2‑
mil
‑
53在30℃条件下的吸附性能,对应hp
‑
mof
‑
1:10:1、hp
‑
mof
‑
1:10:0.8、hp
‑
mof
‑
1:10:0.6、hp
‑
mof
‑
1:10:0.4对stv和ra的总吸附量分别为:168.39mg/g、149.89mg/g、125.39mg/g、38.58mg/g。对应
stv相对于ra的吸附选择性分别为:1.70、1.76、2.11、4.13。
32.图5、图6分别为本实施例所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图及孔径分布图。由图5、图6可见,配体的含量影响缺陷孔的数量。
33.实施例4测试实施例3中所得的hp
‑
mof
‑
1:10:0.6吸附剂对粗糖溶液的吸附性能,在30℃,150rpm,原始粗糖溶液浓度为5mg/ml,其中c
stv
为1.32mg/ml,c
ra
为3.02mg/ml的吸附条件下,其对stv和ra的总吸附量为153.24mg/g。对应stv相对于ra的吸附选择性为2.26。
34.将吸附饱和的hp
‑
mof
‑
1:10:0.6中加入80%的乙醇
‑
水溶液作为解吸液,在150rpm,30℃条件下恒温振荡24h,对吸附甜菊糖苷进行解吸。经过测定,stv和ra的解吸率分别可以达到91.63%和97.58%。由此可见,本发明所制备的hp
‑
nh2‑
mil
‑
53对甜菊糖苷的分离纯化具有良好的循环再生能力和应用能力。
35.实施例5(1)称取2mmol(0.48g)的alcl3·
6h2o,搅拌溶解于40ml dmf中,加入20mmol的癸酸(3.45g)搅拌混合后再加入4mmol(0.72g)的nh2‑
bdc,将混合物搅拌30min后倒入聚四氟乙烯内衬的高压反应釜,在120℃的条件下反应30h,反应结束后将沉淀物用dmf离心清洗三次;(2)将步骤(1)中得到的沉淀物置于40ml含有0.2ml hcl的dmf溶液中,在100℃下搅拌8h完成活化过程,用dmf和甲醇依次清洗三次,并在150℃的真空干燥箱中干燥15h,产物记为hp
‑
mof
‑
1:10:2。bet表征结果显示hp
‑
mof
‑
1:10:2具有730.11m2/g的比表面积,1.07cm3/g的孔体积和0.64~3.67nm的孔径分布范围。
36.采用实施例1中的测试方法测试上述制备得到的hp
‑
mof
‑
1:10:2在30℃条件下的吸附性能,结果显示总吸附量为88.74mg/g,对应stv相对于ra的吸附选择性为1.99。图7、图8分别为本实施例所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图及孔径分布图。
37.实施例6(1)称取2mmol(0.48g)的alcl3·
6h2o,搅拌溶解于40ml dmf中,加入30mmol的癸酸(5.17g)搅拌混合后再加入4mmol(0.72g)的nh2‑
bdc,将混合物搅拌30min后倒入聚四氟乙烯内衬的高压反应釜,在150℃的条件下反应20h,反应结束后将沉淀物用dmf离心清洗三次;(2)将步骤(1)中得到的沉淀物置于40ml含有0.4ml hcl的dmf溶液中,在60℃下搅拌12h完成活化过程,用dmf和甲醇依次清洗三次,并在200℃的真空干燥箱中干燥12h,产物记为hp
‑
mof
‑
1:15:2。bet表征结果表明,hp
‑
mof
‑
1:15:2具有2293.98m2/g的比表面积,1.36cm3/g的孔体积和0.64~2.95nm的孔径分布范围。
38.采用实施例1中的测试方法测试上述制备得到的hp
‑
mof
‑
1:15:2在30℃条件下的吸附性能,结果显示总吸附量为282.52mg/g,对应stv相对于ra的吸附选择性为1.35。图9、图10分别为本实施例所制备的hp
‑
nh2‑
mil
‑
53的n2吸脱附曲线图及孔径分布图。
39.尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1