一种N,N-二甲基-5-羟基色胺的制备方法与流程
一种n,n-二甲基-5-羟基色胺的制备方法
技术领域
1.本发明属于药物化学合成技术领域,尤其涉及一种n,n-二甲基-5-羟基色胺的制备方法。
背景技术:
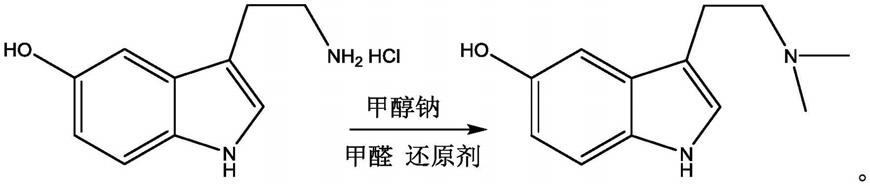
2.n,n-二甲基-5-羟基色胺又名蟾毒色胺,是一种淡黄色固体,最初在蟾蜍中发现,后来也在其他无脊椎动物及一些植物中发现;结构与n,n-二甲基色胺和5-甲氧基-n,n-二甲基色胺类似;在体外与5-ht2a受体结合,与4-溴-2,5-二甲氧基苯基异丙胺(dob)具有类似的亲和力。它既可以作为一种拟精神病药,也可以作为生物标志物用于各种精神疾病的诊断,如精神分裂症和自闭症。
3.n,n-二甲基-5-羟基色胺的合成工艺主要为,第一种合成工艺以5-羟基吲哚为起始原料,经草酰基化、胺化和还原三步反应,得到产品n,n-二甲基-5-羟基基色胺;此工艺存在使用还原剂氢化铝锂,对反应条件及后处理操作要求苛刻,不适于工艺放大,只适合于实验室合成。第二种合成工艺(fischer合成工艺)是以4-甲氧基苯肼盐酸盐为起始原料,在酸催化剂下与n,n-二甲氨基丁醛缩二乙醇进行闭环反应,得到n,n-二甲基-5-羟基色胺,产率70%;这种方法工艺路线虽然短,但原料4-甲氧基苯肼盐酸盐和n,n-二甲氨基丁醛缩二乙醇价格昂贵且不易得到,导致生产成本高,故此合成工艺也不是经济的工艺。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种n,n-二甲基-5-羟基色胺的制备方法,本发明提供的方法制备n,n-二甲基-5-羟基色胺收率高、纯度高。
5.本发明提供了一种n,n-二甲基-5-羟基色胺的制备方法,包括:
6.在含锌催化剂的作用下,将5-羟基色胺盐酸盐溶液、氰基硼氢化钠和甲醛溶液进行反应,然后采用na2co3溶液终止反应,得到n,n-二甲基-5-羟基色胺。
7.优选的,所述含锌催化剂选自三氟醋酸锌和氯化锌中的一种或几种。
8.优选的,所述5-羟基色胺盐酸盐溶液的制备方法包括:
9.将5-羟基色胺盐酸盐和甲醇进行第一混合,得到混合液;
10.将所述混合液和甲醇钠溶液进行第二混合,得到混合物;
11.将所述混合物和醋酸进行第三混合,得到5-羟基色胺盐酸盐溶液。
12.优选的,所述甲醇钠溶液的制备方法包括:
13.将金属钠溶解在甲醇中,得到甲醇钠溶液。
14.优选的,所述混合物的ph值为8~9。
15.优选的,所述5-羟基色胺盐酸盐溶液的ph值为5~6。
16.优选的,所述第一混合在氮气的环境下进行。
17.优选的,所述冰浴冷却的温度为0~5℃。
18.优选的,所述甲醛溶液的质量浓度为35~40%。
19.优选的,所述反应完成后还包括:
20.将得到的反应产物进行过滤,将得到的滤液进行减压蒸馏浓缩。
21.本发明提供了一种合成n,n-二甲基-5-羟基色胺的方法,以5-羟色胺盐酸盐为起始原料,在傅克催化剂(三氟乙酸锌和氯化锌)作用下与甲醛和氰基硼氢化钠反应,得到n,n-二甲基-5-羟基色胺,本发明提供的方法合成工艺简单,合成条件温和,收率高,为95%以上,质量好,含量(纯度)大于99.5%,易于工业化生产。
具体实施方式
22.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员经改进或润饰的所有其它实例,都属于本发明保护的范围。应理解,本发明实施例仅用于说明本发明的技术效果,而非用于限制本发明的保护范围。实施例中,所用方法如无特别说明,均为常规方法。
23.本发明提供了一种n,n-二甲基-5-羟基色胺的制备方法,包括:
24.在含锌催化剂的作用下,将5-羟基色胺盐酸盐溶液、氰基硼氢化钠和甲醛溶液进行反应,然后采用na2co3溶液终止反应,得到n,n-二甲基-5-羟基色胺。
25.在本发明中,n,n-二甲基-5-羟基色胺的制备方法的工艺路线为:
[0026][0027]
在本发明中,所述含锌催化剂优选选自三氟醋酸锌和氯化锌中的一种或几种。
[0028]
在本发明中,所述5-羟基色胺盐酸盐溶液的制备方法优选包括:
[0029]
将5-羟基色胺盐酸盐和甲醇进行第一混合,得到混合液;
[0030]
将所述混合液和甲醇钠溶液进行第二混合,得到混合物;
[0031]
将所述混合物和醋酸进行第三混合,得到5-羟基色胺盐酸盐溶液。
[0032]
在本发明中,所述甲醇优选为无水甲醇。
[0033]
在本发明中,所述5-羟基色胺盐酸盐和甲醇的用量比例优选为1g:(45~50)ml,更优选为1g:(46~48)ml。
[0034]
在本发明中,所述第一混合优选在氮气的环境下进行。
[0035]
在本发明中,所述第一混合优选为搅拌成均一溶液。
[0036]
在本发明中,所述甲醇钠溶液的制备方法优选包括:
[0037]
将金属钠溶解在甲醇中,得到甲醇钠溶液。
[0038]
在本发明中,所述甲醇优选为无水甲醇。
[0039]
在本发明中,所述金属钠和甲醇的用量比例优选为1g:(45~50)ml,更优选为1g:(46~48)ml。
[0040]
在本发明中,所述甲醇钠溶液优选为新制甲醇钠溶液。
[0041]
在本发明中,所述甲醇钠溶液的用量优选使得到混合物的ph值为8~9,更优选为
8.5。
[0042]
在本发明中,所述第二混合优选在冰浴冷却的条件下进行;所述第二混合优选在搅拌的条件下进行。
[0043]
在本发明中,所述冰浴冷却的温度优选为0~5℃,更优选为1~4℃,最优选为2~3℃。
[0044]
在本发明中,所述醋酸的用量优选使得到的5-羟基色胺盐酸盐溶液的ph值为5~6,更优选为5.5。
[0045]
在本发明中,所述甲醛溶液优选为甲醛水溶液;所述甲醛溶液的质量浓度优选为35~40%,更优选为36~38%,最优选为37%。
[0046]
在本发明中,所述5-羟基色胺盐酸盐、氰基硼氢化钠和甲醛的摩尔比优选为1:(1.8~2.2):24,更优选为1:(1.9~2.1):24,最优选为1:2:24。
[0047]
在本发明中,所述反应的温度优选为室温,更优选为20~30℃,最优选为25℃。
[0048]
在本发明中,所述反应优选在搅拌的条件下进行;所述反应的时间优选为2~4小时,更优选为2.5~3.5小时,最优选为3小时。
[0049]
在本发明中,所述终止反应之前优选还包括:
[0050]
取得到的反应液进行检测,当5-羟基色胺≤0.5wt%时终止反应。
[0051]
在本发明中,所述检测的方法优选为液相色谱检测;所述液相色谱检测采用的仪器优选为高效液相色谱仪lc-2030c 3d plus;采用的检测器优选为uv检测器;采用的色谱柱优选为c18 250mm*4.6mm,5μm;采用的流动相优选为0.1mmol/l磷酸盐水溶液:甲醇=88:12;采用的波长优选为220nm;采用的进样量优选为10μl;采用的流速优选为1ml/min;出峰时间优选为5.685min。
[0052]
在本发明中,所述na2co3溶液优选为na2co3水溶液;所述na2co3溶液的浓度优选为1.5~2.5mol/l,更优选为1.8~2.2mol/l,最优选为2mol/l。
[0053]
在本发明中,所述终止反应优选为滴加na2co3溶液使反应体系的ph值为8~9,更优选为8.5。
[0054]
在本发明中,所述反应完成后优选还包括:
[0055]
将得到的反应产物进行过滤,将得到的滤液进行减压蒸馏浓缩。
[0056]
在本发明中,所述减压蒸馏浓缩的真空度优选为-0.095~-0.097mpa,更优选为-0.096mpa;水浴温度优选为45~50℃,更优选为46~48℃;所述减压蒸馏过程中蒸馏出甲醇水,待蒸馏气相温度最高达到45℃时,停止减压蒸馏。
[0057]
在本发明中,所述减压蒸馏后优选还包括:
[0058]
将得到的减压蒸馏产品进行萃取,合并萃取液。
[0059]
在本发明中,所述萃取过程中优选将得到的减压蒸馏产品加冰水稀释。
[0060]
在本发明中,所述萃取优选采用乙酸乙酯萃取,所述萃取的次数优选为2~4次,更优选为3次。
[0061]
在本发明中,所述合并萃取液优选为合并乙酸乙酯萃取液。
[0062]
在本发明中,所述萃取完成后优选还包括:
[0063]
将合并后的萃取液再次进行减压蒸馏,得到n,n-二甲基-5-羟基色胺。
[0064]
在本发明中,所述再次进行减压蒸馏的真空度优选为-0.095~-0.097mpa,更优选
plus;检测器:uv检测器;色谱柱:c18 250mm*4.6mm,5μm;流动相:0.1mmol/l磷酸盐水溶液:甲醇=80:20;波长:220nm;进样量:10μl;流速:1ml/min;出峰时间:8.523min。
[0075]
根据纯度计算产率。
[0076]
实施例2
[0077]
在氮气环境下,将21.46克(折纯为21.25g)(0.1mol)5-羟基色胺盐酸盐加入到1000毫升无水甲醇中,搅拌下成均一溶液;冰浴冷却到0~5℃,搅拌下加入由2.43g金属钠溶解在120ml无水甲醇新制的甲醇钠溶液调ph=8~9,接着加入23.4毫升醋酸(0.4mol)调节ph=5~6,再加入催化剂0.54g氯化锌和12.56g氰基硼氢化钠(0.2mol),最后滴加质量浓度为37%甲醛水溶液0.180l(2.4mol),滴加完毕,回升温度到室温,继续搅拌3h,取样检测,5-羟基色胺含量为0.5wt%(检测方法同实施例1),滴加2mol/l的na2co3溶液调ph约8~9终止反应,过滤,滤液减压蒸馏浓缩:
[0078]
开启真空装置,保持真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出甲醇水,待蒸馏气相温度最高达到45℃,停止减压蒸馏,关闭真空;加入2500毫升冰水稀释,用3000毫升乙酸乙酯萃取,共萃取两次,合并乙酸乙酯萃取液,浓缩除尽溶剂(减压蒸馏:真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出乙酸乙酯,待蒸馏气相温度最高达到40℃,停止减压蒸馏),得到18.48g产品;产率90.20%,含量(纯度)99.56%。
[0079]
实施例3
[0080]
在氮气环境下,将21.46克(折纯为21.25g)(0.1mol)5-羟基色胺盐酸盐加入到1000毫升无水甲醇中,搅拌下成均一溶液;冰浴冷却到0~5℃,搅拌下加入由2.43g金属钠溶解在120ml无水甲醇新制的甲醇钠溶液调ph=8~9,接着加入23.4毫升醋酸(0.4mol)调节ph=5~6,再加入催化剂0.54g三氟乙酸锌和11.93g氰基硼氢化钠(0.19mol),最后滴加质量浓度37%甲醛水溶液0.180l(2.4mol),滴加完毕,回升温度到室温,继续搅拌3h,取样检测,5-羟基色胺含量为0.82wt%(检测方法同实施例1),滴加2mol/l的na2co3溶液调ph约8~9终止反应,过滤,滤液减压蒸馏浓缩:
[0081]
开启真空装置,保持真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出甲醇水,待蒸馏气相温度最高达到45℃,停止减压蒸馏,关闭真空;加入2500毫升冰水稀释,用3000毫升乙酸乙酯萃取,共萃取两次,合并乙酸乙酯萃取液,浓缩除尽溶剂(减压蒸馏:真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出乙酸乙酯,待蒸馏气相温度最高达到40℃,停止减压蒸馏),得到19.16g产品;产率93.50%,含量(纯度)99.55%。
[0082]
实施例4
[0083]
在氮气环境下,将21.46克(0.1mol)(折纯为21.25g)5-羟基色胺盐酸盐加入到1000毫升无水甲醇中,搅拌下成均一溶液;冰浴冷却到0~5℃,搅拌下加入由2.43g金属钠溶解在120ml无水甲醇新制的甲醇钠溶液调ph=8~9,接着加入23.4毫升醋酸(0.4mol)调节ph=5~6,再加入催化剂0.54g三氟乙酸锌和13.19g氰基硼氢化钠(0.21mol),最后滴加质量浓度为37%甲醛水溶液0.180l(2.4mol),滴加完毕,回升温度到室温,继续搅拌3h,取样检测,5-羟基色胺含量为0.45wt%(检测方法同实施例1),滴加2mol/l的na2co3溶液调ph约8~9终止反应,过滤,滤液减压蒸馏浓缩:
[0084]
开启真空装置,保持真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出甲醇水,待蒸馏气相温度最高达到45℃,停止减压蒸馏,关闭真空;加入2500毫升冰水
稀释,用3000毫升乙酸乙酯萃取,共萃取两次,合并乙酸乙酯萃取液,浓缩除尽溶剂(减压蒸馏:真空度-0.095~-0.097mpa,水浴温度设定45~50℃,减压蒸馏出乙酸乙酯,待蒸馏气相温度最高达到40℃,停止减压蒸馏),得到19.47g产品;产率为95.03%,含量(纯度)99.57%。
[0085]
本发明提供了一种合成n,n-二甲基-5-羟基色胺的方法,以5-羟色胺盐酸盐为起始原料,在傅-克催化剂(三氟乙酸锌和氯化锌)作用下与甲醛和氰基硼氢化钠反应得到n,n-二甲基-5-羟基色胺;本发明提供的方法合成工艺简单,合成条件温和;加入傅-克催化剂三氟醋酸锌或氯化锌,优化出反应条件,在0~5℃滴加反应物料,滴加完毕,回升温度到室温,反应3小时;产品收率高,为90%以上,质量好,含量大于99.5%。
[0086]
以上所述的仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1