一种特西他赛C-13侧链的合成方法
一种特西他赛c-13侧链的合成方法
技术领域
1.本发明涉及一种特西他赛c-13侧链的合成方法,属于有机化学技术领域。
背景技术:
2.紫杉醇是一种天然产物抗癌药物,由美国化学家沃尔和瓦尼在1966年首次从西洋红豆杉树皮中首次提取出,对卵巢癌,肺癌及乳腺癌的恶性肿瘤都有很好治疗效果。其中紫杉醇,多西他赛和卡巴他赛是这类化合物中最典型的代表药物制剂,然而患者在注射紫杉醇药物和多西他赛药物后均会有一些不良的药物反应,尤其是引起患者对药物神经过敏反应,甚至偶尔会导致患者死亡,同时这些药物还会引起白细胞降低以及神经功能损伤等其它一些不良反应。
3.特西他赛是一种新型可口服紫杉烷类抗癌药物,不仅有高的抗癌活性,而且作为口服胶囊,避免患者对药物注射产生副反应,提高了患者用药的便利性。特西他赛药品可以单独使用或与其他药品联用时,可以改变特西他赛的用量,这样可以对患者的治疗周期做出灵活的调整。2008年美国食品药品管理局批准genta公司将特西他赛做为治疗晚期黑素癌的紫杉烷类口服试剂,目前已经在进行三期临床研究中。现有特西他赛化合物都是通过具有巴卡丁结构的特西他赛母环和其相应的侧链对接制得的,因此寻找一条适合切实可行的合成特西他赛(tesetaxel)的路线变得非常重要。所以开发出一条高效合成特西他赛c-13侧链的路线,对特西他赛的合成与规模制备具有重要的意义。
4.现有特西他赛侧链的主要合成方法:
5.2002年由日本第一制药株式会社的tsunehiko soga,kouichi uoto和yasuyuki takeda报道的特西他赛半合成路线中侧链合成路线。以3-氟吡啶-2-醛为原料与ch3coch2cocl发生staudinger反应合成β-内酰胺,对羟基和氮上保护基处理,经过手性柱拆分得到光学纯的β-内酰胺形式的特西他赛侧链前体。
[0006][0007]
2006年由日本第一制药株式会社的sato kouji,yagi tsutomu和kitagawa yutaka报道的ep 1375495 b1专利中特西他赛半合成路线中侧链合成路线。以3-氟吡啶-2-醛为原料合成β-内酰胺,由脂肪酶水解拆分出得到需要的光学纯的β-内酰胺,对羟基和氮上保护基处理得到目标产物特西他赛侧链前体。
[0008][0009]
2012年由真他公司john k.thottathil和raymond p.warrell报道的us 2012/0220634a1专利中特西他赛半合成路线中侧链合成路线。
[0010]
技术实现要素:
[0011]
本发提出了一种用合成路线短,效率高的合成β-内酰胺形式的侧链前体,再由脂肪酶拆分得到光学纯的特西他赛侧链前体,省去羟基脱除保护基和加保护基的两步,缩短了路线步骤。
[0012]
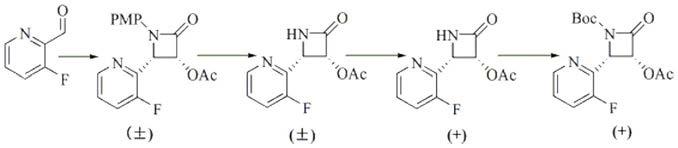
本发明的技术方案是:一种特西他赛c-13侧链的合成方法。其特征是包括以下步骤:
[0013]
(1)以3-氟吡啶-2-醛和对甲氧基苯胺为初始原料,通过缩合反应合成席夫碱。
[0014]
(2)席夫碱与乙酰氧基乙酰氯之间发生staudinger反应合成具有α-乙酰氧基取代的β-内酰胺。
[0015]
(3)用硝酸铈铵试剂将β-内酰胺的氮原子上对甲氧基苯基保护基脱去。
[0016]
(4)用牛肝酶水解拆分脱去保护基的β-内酰胺,得到具光学纯的手性β-内酰胺。
[0017]
(5)用二碳酸叔丁酯(boc2o)在催化量的4-二甲氨基吡啶下反应,对手性β-内酰胺上氮原子进行保护,得到特细他赛的c-13的侧链前体。
[0018]
上述合成方法具体包括以下步骤:
[0019]
(1)羟基乙酸溶于1,4-二氧六环中,加入乙酰氯升温至90-100℃,反应3-5小时至无气体产生,然后升温至115-130℃蒸出未反应的乙酰氯和1,4-二氧六环溶剂,得到乙酰氧基乙酸,加入草酰氯和少量dmf,在室温下反应1-3小时后加入草酰氯,继续反应1-3小时至无气体产生,制得到乙酰氧基乙酰氯。
[0020][0021]
(2)3-氟吡啶-2-甲醛与对甲氧基苯胺溶于苯中,加入无水硫酸钠,常温下搅拌0.5-2小时后经过滤、旋干、真空干燥约2-4小时;在无水无氧氩气保护条件下将得到产物溶于二氯甲烷中,-80
‑‑
70℃下加入三乙胺,缓慢滴加乙酰氧基乙酰氯,室温下加入水淬灭反应,用乙酸乙酯萃取、饱和nacl溶液洗涤、无水硫酸钠干燥、过滤、旋干后得到β-内酰胺粗产物,用二氯甲烷-石油醚重结晶得到高纯β-内酰胺。
[0022][0023]
(3)将β-内酰胺溶于乙腈中,在-15-0℃下缓慢加入硝酸铈铵,反应20-50分钟,用饱和碳酸氢钠溶液淬灭反应。用乙酸乙酯萃取,饱和氯化钠溶液洗涤,无水硫酸钠干燥,经旋干、分离得到脱pmp保护基的外消旋β-内酰胺。
[0024][0025]
(4)将外消旋β-内酰胺溶于适量的乙醚中,在一定温度下加入牛肝酶溶液,通过tlc检测反应,当反应结束后加入丙酮淬灭反应。用乙酸乙酯萃取,饱和nacl溶液洗涤,无水硫酸钠干燥,经旋干、柱层析分离后得到手性β-内酰胺。
[0026][0027]
(5)手性β-内酰胺溶于二氯甲烷中,加入boc2o和dmap后反应20-50分钟,直接进行柱层析分离得到c-13的侧链前体。
[0028]
具体实施方式:
[0029]
以下结合实施例对本发明进一步说明,但是本发明并不局限于此。
[0030]
实施例1
[0031]
(1)90g羟基乙酸溶于600ml 1,4-二氧六环中,加入80ml乙酰氯升温至90℃,反应3小时至无气体产生,然后升温至120℃蒸出未反应的乙酰氯和1,4-二氧六环溶剂,得到乙酰氧基乙酸,将乙酰氧基乙酸溶于500ml苯中,加入170ml草酰氯和几滴dmf,在室温下反应1小时后加入84ml草酰氯,继续反应1.5小时至无气体产生,制得到乙酰氧基乙酰氯。
[0032]
(2)15g 3-氟吡啶-2-甲醛与14.8g对甲氧基苯胺溶于200ml苯中,加入无水硫酸钠15g,常温下搅拌0.5小时后经过滤、旋干、真空干燥约2.5小时;在无水无氧氩气保护条件下将得到产物溶于100ml二氯甲烷中,-80℃下加入66ml三乙胺,缓慢滴加65g乙酰氧基乙酰氯,室温下加入水淬灭反应,用200ml乙酸乙酯萃取三次,饱和nacl溶液洗涤两次,无水硫酸钠干燥,过滤,旋干,将得到粗产物用二氯甲烷-石油醚重结晶,得到高纯β-内酰胺。
[0033]
(3)将10gβ-内酰胺溶于乙腈中,在-15℃下缓慢加入60g硝酸铈铵,反应20分钟。用饱和碳酸氢钠溶液淬灭反应。用300ml乙酸乙酯萃取五次,用饱和氯化钠溶液洗涤两次,无水硫酸钠干燥,旋干,柱层析分离。得到得到脱pmp保护基的外消旋β-内酰胺产率。
[0034]
(4)30g用绞肉机搅碎过的牛肝加入ph=8的磷酸氢钾和磷酸二氢钾缓冲液,配成100ml牛肝溶液。将外消旋β-内酰胺溶于适量的乙醚中,在一定温度下加入牛肝酶溶液,通过tlc检测反应,当反应结束后加入丙酮淬灭反应。用乙酸乙酯萃取,饱和nacl溶液洗涤,无水硫酸钠干燥,经旋干、柱层析分离后得到手性β-内酰胺。
[0035]
(5)手性β-内酰胺溶于二氯甲烷中,加入1当量boc2o,0.2当量dmap,反应20分钟,直接上柱子进行柱层析分离c-13的侧链前体。
[0036]
实施例2
[0037]
(1)90g羟基乙酸溶于600ml 1,4-二氧六环中,加入80ml乙酰氯升温至90℃,反应3.5小时至无气体产生,然后升温至120℃蒸出未反应的乙酰氯和1,4-二氧六环溶剂,得到乙酰氧基乙酸,将乙酰氧基乙酸溶于500ml苯中,加入170ml草酰氯和几滴dmf,在室温下反应1.5小时后加入80ml草酰氯,继续反应1.5小时至无气体产生,制得到乙酰氧基乙酰氯。
[0038]
(2)10g 3-氟吡啶-2-甲醛与13g对甲氧基苯胺溶于200ml苯中,加入无水硫酸钠15g,常温下搅拌0.5小时后经过滤、旋干、真空干燥约2.5小时;在无水无氧氩气保护条件下将得到产物溶于100ml二氯甲烷中,-80℃下加入66ml三乙胺,缓慢滴加60g乙酰氧基乙酰氯,室温下加入水淬灭反应,用200ml乙酸乙酯萃取三次,饱和nacl溶液洗涤两次,无水硫酸钠干燥,过滤,旋干,将得到粗产物用二氯甲烷-石油醚重结晶,得到高纯β-内酰胺。
[0039]
(3)将10gβ-内酰胺溶于乙腈中,在-15℃下缓慢加入60g硝酸铈铵,反应20分钟。用饱和碳酸氢钠溶液淬灭反应。用乙酸乙酯萃取五次,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋干,柱层析分离。得到得到脱pmp保护基的外消旋β-内酰胺产率。
[0040]
(4)40g用绞肉机搅碎过的牛肝加入ph=8的磷酸氢钾和磷酸二氢钾缓冲液,配成100ml牛肝溶液。将外消旋β-内酰胺溶于适量的乙醚中,在一定温度下加入牛肝酶溶液,通过tlc检测反应,当反应结束后加入丙酮淬灭反应。用乙酸乙酯萃取,饱和nacl溶液洗涤,无水硫酸钠干燥,经旋干、柱层析分离后得到手性β-内酰胺。
[0041]
(5)手性β-内酰胺溶于二氯甲烷中,加入1当量boc2o,0.2当量dmap,反应20分钟,直接上柱子进行柱层析分离c-13的侧链前体。
[0042]
实施例3
[0043]
(1)70g羟基乙酸溶于300ml 1,4-二氧六环中,加入80ml乙酰氯升温至97℃,反应
3.5小时至无气体产生,然后升温至120℃蒸出未反应的乙酰氯和1,4-二氧六环溶剂,得到乙酰氧基乙酸,将乙酰氧基乙酸溶于500ml苯中,加入170ml草酰氯和几滴dmf,在室温下反应1.5小时后加入80ml草酰氯,继续反应2小时至无气体产生,制得到乙酰氧基乙酰氯。
[0044]
(2)10g 3-氟吡啶-2-甲醛与13g对甲氧基苯胺溶于200ml苯中,加入无水硫酸钠15g,常温下搅拌0.5小时后经过滤、旋干、真空干燥约2.5小时;在无水无氧氩气保护条件下将得到产物溶于100ml二氯甲烷中,-80℃下加入66ml三乙胺,缓慢滴加47g乙酰氧基乙酰氯,室温下加入水淬灭反应,用200ml乙酸乙酯萃取,饱和nacl溶液洗涤,无水硫酸钠干燥,过滤,旋干,将得到粗产物用二氯甲烷-石油醚重结晶,得到高纯β-内酰胺。
[0045]
(3)将10gβ-内酰胺溶于乙腈中,在-10℃下缓慢加入60g硝酸铈铵,反应20分钟。用饱和碳酸氢钠溶液淬灭反应。用乙酸乙酯萃取,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋干,柱层析分离。得到得到脱pmp保护基的外消旋β-内酰胺。
[0046]
(4)20g用绞肉机搅碎过的牛肝加入ph=8的磷酸氢钾和磷酸二氢钾缓冲液,配成100ml牛肝溶液。将外消旋β-内酰胺溶于适量的乙醚中,在一定温度下加入牛肝酶溶液,通过tlc检测反应,当反应结束后加入丙酮淬灭反应。用乙酸乙酯萃取,饱和nacl溶液洗涤,无水硫酸钠干燥,经旋干、柱层析分离后得到手性β-内酰胺。
[0047]
(5)手性β-内酰胺溶于二氯甲烷中,加入1当量boc2o,0.2当量dmap,反应20分钟,直接上柱子进行柱层析分离c-13的侧链前体。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1