一种高纯度丁位内酯的制备方法与流程
1.本发明属于香精香料领域,具体地,涉及一种高纯度丁位内酯的制备方法。
背景技术:
2.丁位内酯又称为δ-内酯,主要用于食用香精中,也可少量添加到某些化妆品香精和香水中,可起到画龙点睛的作用,δ-内酯的香气和香味与相应的γ-内酯相比更为柔和,在配方中往往起到关键性的作用。例如,δ-十二内酯对于白脱、冰淇淋和乳酪香精配方中是不可缺少的重要成分,天然的δ-内酯存在于桃、椰子、覆盆子、芦笋等中,有些也存在于牛奶、奶油及肉类中;多具有奶油香气,以及椰子等水果香味。由于δ-内酯比相同碳数的γ-内酯更难合成,因此,它的合成一直以来受到广泛的重视。目前主要的合成方法是:先利用脂肪醛与环戊酮在碱性条件下羟醛缩合,再加氢合成2-烷基环戊酮,然后经拜耳-维利格(baeyer-villiger)氧化得到δ-内酯。
3.赵延伟等在《上海应用技术学院学报》(自然科学版)上报道,以戊二酸酐为起始原料,经溴戊烷的格氏试剂加成反应,得到5-氧代癸酸,然后经硼氢化钠还原再酸化得到丁位癸内酯,总收率72%。但是此方法格氏试剂制备条件苛刻且不易保存,操作难度大,难以实现工业化生产。
4.2012年安徽理工大学化学工程学院李广学、姜丰等人在第29卷第5期的《精细石油化工报刊》提出来一种合成δ-癸内酯的合成方法:以正戊醛和环戊酮经羟醛缩合、脱水反应合成戊烯基环戊酮,经加氢制得2-戊基环戊酮,2-戊基环戊酮在过氧乙酸作用下经baeyer-villiger氧化合成δ-癸内酯,收率为61.2%。该方法收率高且产品香气满足市场要求,但是由于过氧乙酸性质活泼且易爆炸,在工业生产中存在严重的安全隐患,同时该方法有大量的乙酸废水生成,不利于环保要求。
5.中国专利cn1962653a公开了一种δ-十一内酯的合成方法,在室温的条件下,以甲酸为溶剂,2-己基环戊酮经尿素-过氧化氢复合物氧化得到粗品δ-十一内酯,然后用真空薄膜蒸馏的方法获得纯品。此专利中由于过氧化物性质活泼且易爆炸,在工业生产中存在严重的安全隐患,同时该方法有大量的废水生成,不利于环保要求。
技术实现要素:
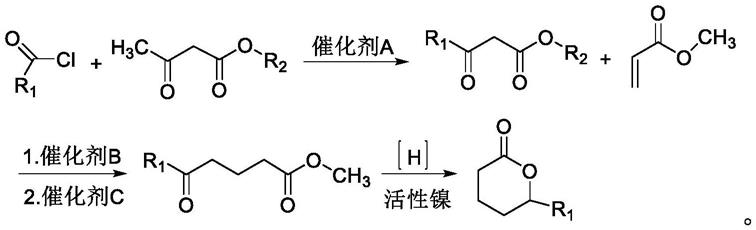
6.本发明的目的在于提供一种安全、环保且具有较高得率的高纯度δ-内酯的合成方法,以乙酰基乙酸酯、烷基酰氯和烯酸甲酯为原料,逐步反应获得丁位内酯,以解决现有丁位内酯生产方法中存在的安全隐患和环保问题。
7.本发明的目的可以通过以下技术方案实现:
8.一种高纯度丁位内酯的制备方法,包括以下步骤:
9.a.以二氯甲烷为溶剂,烷基酰氯和乙酰基乙酸酯为原料在催化剂a作用下反应得到烷基酰基乙酸酯中间体;
10.b.烷基酰基乙酸酯中间体在碱性催化剂b的作用下与丙烯酸甲酯进行迈克尔加成
合成烷基酰基戊二酸酯;
11.c.烷基酰基戊二酸酯在酸性催化剂c的条件下加热脱羧得到烷基酰基丁酸酯;
12.d.以活性镍为催化剂催化下,烷基酰基丁酸酯在一定压力和温度条件下加氢、环化得到δ-内酯。
13.进一步地,步骤a中乙酰基乙酸酯为乙酰基乙酸甲酯和乙酰基乙酸乙酯中的一种,优选乙酰基乙酸乙酯。
14.进一步地,步骤a中催化剂a为氯化镁、氯化钙、氢氧化钙和氧化钙中的一种,优选氯化镁;乙酰基乙酸酯和氯化镁的摩尔比为1.0:0.5-1.5,优选1:1。
15.进一步地,步骤a中烷基酰氯、乙酰基乙酸酯的摩尔比为1.0-2.0:1.0,优选1.3:1。
16.进一步地,步骤a中烷基酰氯中烷基的碳原子数目n=1-20。
17.进一步地,步骤a中反应温度为0-20℃。
18.进一步地,步骤b中碱性催化剂b为氢氧化钠、氢氧化钾、碳酸钠和碳酸钾中的一种,优选碳酸钾,以丙烯酸甲酯的物质的量计算,碳酸钾的用量为5-15%mol。
19.进一步地,步骤b中反应温度为40-50℃。
20.进一步地,步骤c中酸性催化剂c为硫酸、盐酸、硼酸中的一种,优选硼酸,以烷基酰基戊二酸酯物质的量计算硼酸的用量为100-200%mol,优先100%mol。
21.一种高纯度丁位内酯的制备方法,其合成路线如下所示:
[0022][0023]
本发明的有益效果:
[0024]
1)原料试剂皆为工业基础原料,价格低廉,降低了生产成本;
[0025]
2)反应条件简单,易于工业化,同时生产过程中不产生易爆物质和大量的废酸,生产安全性较高,且环保;
[0026]
3)因本发明中不含氧化工艺,使得最终产物没有同分异构体,纯度高达99.9%。
[0027]
本发明的优点在于合成的原料价格低廉、收率较高、产品没有异构体纯度高且反应条件简单,易于工业化生产。
具体实施方式
[0028]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0029]
实施例1
[0030]
己酰基乙酸甲酯的制备:
[0031]
在带有搅拌、温度计和滴液漏斗的2000ml三口烧瓶中加入二氯甲烷1000ml,无水氯化镁95.2g(1mol),搅拌下加入乙酰基乙酸甲酯116.1g(1mol),冷却至0℃以下,保持温度滴入己酰氯148g(1.1mol),反应30分钟,0℃反应2小时,冷却到0℃以下,加入预冷5%盐酸500ml,搅拌20分钟,分出有机层,水相用300ml二氯甲烷萃取两次,合并有机相,用5%盐酸200ml洗涤一次,有机相用无水硫酸镁干燥,常压回收溶剂二氯甲烷,剩余物溶于450ml乙醚中,室温,搅拌下滴入10%氨水400ml,反应1小时,分出醚层,乙醚100ml/次萃取水相2次,合并有机相,加入5%盐酸100ml,室温搅拌1小时,调节ph=2.1,分出有机相,乙醚100ml/次萃取水相2次,合并有机相,用饱和碳酸氢钠洗涤,常压蒸出乙醚,采用分馏柱减压收集馏分,得到己酰基乙酸甲酯131.3克,产率61%,纯度80%(gc)。
[0032]
实施例2
[0033]
己酰基乙酸甲酯的制备:
[0034]
在带有搅拌、温度计和滴液漏斗的2000ml三口烧瓶中加入二氯甲烷1000ml,无水氯化镁95.2g(1mol),搅拌下加入乙酰基乙酸甲酯116g(1mol),冷却至0℃以下,保持温度滴入己酰氯174.9g(1.3mol),反应30分钟,20℃反应2小时,冷却到0℃以下,加入预冷5%盐酸600ml,搅拌20分钟,分出有机层,水相用300ml二氯甲烷萃取两次,合并有机相,用5%盐酸200ml洗涤一次,有机相用无水硫酸镁干燥,常压回收溶剂二氯甲烷,剩余物溶于450ml乙醚中,室温,搅拌下滴入10%氨水450ml,反应1小时,分出醚层,乙醚100ml/次萃取水相2次,合并有机相,加入5%盐酸100ml,室温搅拌1小时,调节ph=3,分出有机相,乙醚100ml/次萃取水相2次,合并有机相,用饱和碳酸氢钠洗涤,常压蒸出乙醚,采用分馏柱减压收集馏分,得到己酰基乙酸甲酯124.9克,产率66%,纯度91%(gc)。
[0035]
实施例3
[0036]
己酰基丁酸甲酯的制备:
[0037]
在配有机械搅拌,温度计和滴液漏斗的1000ml三口烧瓶中加入实施例1中的己酰基乙酸甲酯124.9g(gc:80%,0.66mol)和氢氧化钠固体1.32g(0.033mol)开启搅拌升温至40度,待釜温稳定后,开始缓慢滴加丙烯酸甲酯73.78g(0.858mol),约4小时滴加结束,控制滴加过程中温度为40-45℃,滴加结束后40℃保温两小时,降温至室温,加3.17g(0.033mo)的38%盐酸中和,200g饱和盐水水洗一次,油层水泵减压回收过量丙烯酸甲酯,得到釜内物料151.36g(gc:90%收率80%),重新搭建常压蒸流装置,瓶内加入151.36g(0.528mol)2-己酰基戊二酸甲酯粗品和32.2g(0.528mol)硼酸。缓慢搅拌升温至150℃且有明显回流,及时蒸出低沸点液体,搅拌反应8小时后取样做gc分析原料反应完全为止,然后降温至20℃,滴加10%氢氧化钠溶液调节釜内物料ph呈中性,油层经减压粗蒸得到己酰基丁酸甲酯粗品105.6g,gc分析含量90%,两步总收率为72%,留待下一步使用。
[0038]
实施例4
[0039]
己酰基丁酸甲酯的制备:
[0040]
在配有机械搅拌,温度计和滴液漏斗的1000ml三口烧瓶中加入实施例2中的己酰基乙酸甲酯124.9g(gc:91%,0.66mol)和碳酸钾固体5.93g(0.043mol)开启搅拌升温至40度,待釜温稳定后,开始缓慢滴加丙烯酸甲酯73.78g(0.858mol),约4小时滴加结束,控制滴加过程中温度为45-50℃,滴加结束后50℃保温两小时,降温至室温,加8.26g(0.086mol)的38%盐酸中和,200g饱和盐水水洗一次,油层水泵减压回收过量丙烯酸甲酯,得到釜内物料
170.2g(gc:90%收率90%),重新搭建常压蒸流装置,釜内加入170.2g(0.594mol)2-己酰基戊二酸甲酯和36.23g(0.594mol)硼酸。缓慢搅拌升温至150℃且有明显回流,及时蒸出低沸点液体,搅拌反应8小时后取样做gc分析原料反应完全为止,然后降温至20℃,滴加10%氢氧化钠溶液调节釜内物料ph呈中性,油层经减压粗蒸得到己酰基丁酸甲酯粗品117.5g,gc分析含量91%,两步总收率81%,留待下一步使用。
[0041]
实施例5
[0042]
δ-癸内酯的制备:
[0043]
在500ml的高压加氢釜内加入实施例3中的己酰基丁酸甲酯粗品117.5g(gc:91%0.53mol),3g雷尼镍催化剂,氮气置换空气,氢气置换氮气,充入氢气至0.8mpa,搅拌下加热至120℃。继续搅拌反应至压力不再下降,约需12小时。冷却到室温,滤出催化剂。滤液减压蒸馏收集108.3gδ-癸内酯粗品馏分,gc分析含量93%含量,收率95%。粗品经减压精馏得到δ-癸内酯成品90.6g,纯度99.8%。三步反应总收率约为50.8%。
[0044]
实施例6
[0045]
δ-癸内酯的制备:
[0046]
与实施例5相比,加入的己酰基丁酸甲酯为实施例4制备的,其余步骤相同,其中,109.6gδ-癸内酯粗品馏分,gc分析含量94%,收率95.4%。粗品经减压精馏得到δ-癸内酯成品91.2g,纯度99.3%。三步反应总收率约为51.2%。
[0047]
实施例7
[0048]
丙酰基乙酸甲酯的制备:与实施例1相比,将己酰氯替换成丙酰氯,且丙酰氯的加入量为1.1mol,其余相同,得到丙酰基乙酸甲酯96.06克,产率62%,纯度84%(gc)。
[0049]
实施例8
[0050]
丙酰基丁酸甲酯的制备:与实施例3相比,将丙酰基乙酸甲酯替换成实施例7制备的丙酰基乙酸甲酯(96.06g,0.62mol)),其余相同,得釜内物料119.04g(gc:90%收率82%);然后将釜内料和(30.64g)36.23g((0.495)0.594mol)硼酸混合,缓慢搅拌升温至150℃且有明显回流,及时蒸出低沸点液体,搅拌反应8小时后取样做gc分析原料反应完全为止,然后降温至20℃,滴加10%氢氧化钠溶液调节釜内物料ph呈中性,油层经减压粗蒸得到丙酰基丁酸甲酯粗品(87.3g)117.5g,gc分析含量91%,两步总收率81%,留待下一步使用。
[0051]
实施例9
[0052]
丁位庚内酯的制备:与实施例5相比,己酰基丁酸甲酯粗品替换成实施例8制备的丙酰基丁酸甲酯,其余相同,得丁位庚内酯91.2g,纯度收率95%;三步反应总收率约为47.71%。
[0053]
实施例10
[0054]
丁位辛内酯的制备:
[0055]
步骤一、同实施例1相比,将己酰氯替换成丁酰氯,且丁酰氯的加入量为1.1mol,得到丁酰基乙酸甲酯80.4(116.38)克,产率61.8%,纯度84%(gc);
[0056]
步骤二、与实施例3相比,将己酰基乙酸甲酯替换成步骤一制备的丁酰基乙酸甲酯,其余相同,得釜内物料(122.33g)170.2g(gc:92%收率81.2%);然后将釜内料和31.03g(0.502mol)硼酸混合,缓慢搅拌升温至150℃且有明显回流,及时蒸出低沸点液体,搅拌反应8小时后取样做gc分析原料反应完全为止,然后降温至20℃,滴加10%氢氧化钠溶液调节
釜内物料ph呈中性,油层经减压粗蒸得到丁酰基丁酸甲酯粗品(91.21g)117.5g,gc分析含量93%,两步总收率79.8%,留待下一步使用
[0057]
步骤三、与实施例5相比,己酰基丁酸甲酯粗品替换成步骤三制备的丁酰基丁酸甲酯,其余相同,得67g丁位辛内酯,纯度99.5%,三步反应总收率约为51.2(46.85)%。
[0058]
实施例11
[0059]
丁位十二内酯的制备:
[0060]
步骤一、同实施例1相比,将己酰氯替换成辛酰氯,且辛酰氯的加入量为1.1mol,得到辛酰基乙酸甲酯148.35g,产率60%,纯度81%(gc);
[0061]
步骤二、与实施例3相比,将己酰基乙酸甲酯替换成步骤一制备的辛酰基乙酸甲酯,其余相同,得釜内物料170.2(158.31)g(gc:89%收率82%);然后将釜内料和30.42g(0.492mol)硼酸混合,缓慢搅拌升温至150℃且有明显回流,及时蒸出低沸点液体,搅拌反应8小时后取样做gc分析原料反应完全为止,然后降温至20℃,滴加10%氢氧化钠溶液调节釜内物料ph呈中性,油层经减压粗蒸得到辛酰基丁酸甲酯粗品124.7g,gc分析含量89%,两步总收率83%,留待下一步使用
[0062]
步骤三、与实施例5相比,辛酰基丁酸甲酯粗品替换成步骤三制备的丁酰基丁酸甲酯,其余相同,得91.8g丁位十二内酯,纯度99.8%(收率:95%),三步反应总收率约为(47.3%)。
[0063]
在说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
[0064]
以上内容仅仅是对本发明所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离发明或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1