一种具有多发光单元的红光材料及其制备方法和应用与流程
1.本发明涉及有机电致发光技术领域,主要涉及一种具有多发光单元的红光材料及其制备方法和应用。
背景技术:
2.1987年,美国kodak公司的c.w.tang等人制备了以氧化铟锡(ito)和金属合金薄膜分别为阳极和阴极,以芳香胺类材料作为空穴传输层,以8
‑
羟基喹啉的铝配合物(alq3)作为电子传输层与发光层的发光器件,器件效率为1.51lm/w(见c.w. tang and s. a. wanslyke, appl. phys. lett., 1978, 51, 913),从此全球掀起了对于有机电致发光技术的研究。
3.根据电子自旋守恒的量子力学跃迁规律的约束,传统荧光染料只能够利用25%的单线态激子的能量,内量子效率的极限是25%。1998年美国普林斯顿大学的forrest等人利用金属铂配合物磷光材料制备出内量子效率23%、外量子效率4%的发光器件(见m.a.baldo, d.f.o’brienetal, nature, 1998, 395, 151)。铱、铂等重金属诱导的自旋轨道耦合作用使得三线态激子可以直接通过辐射跃迁过程回到基态发射磷光,其理论内量子效率可以达到100%。但是铱铂等重金属的引入提高了磷光材料的成本,而且深蓝光磷光材料的化学稳定性差,器件在高亮度下的效率滚降严重。因此急需开发廉价稳定的有机小分子材料实现高效稳定的oled器件。
4.近年来,热活化延迟荧光(tadf: thermally activated delayed fluorescence)材料由于具有三线态激子可以反系间窜越过程回到单线态,进而发射延迟荧光的特点,同样可以实现100%的激子利用率(见h.uoyama, k.goushi, k.shizu, h.nomura, c.adachi, nature, 2012, 492, 234)。近年来天蓝光、绿光等发光波长的tadf材料获得了飞速的发展(见 t. a. lin, t. chatterjee, w. l. tsai, w. k. lee, m. j. wu, m. jiao, k. c. pan, c. l. yi, c. l. chung, k. t. wong, and c. c. wu, adv. mater. , 2016, 28, 6976 ; tien
‑
lin wu, min
‑
jie huang, chih
‑
chun lin, pei
‑
yun huang, tsu
‑
yu chou, ren
‑
wu chen
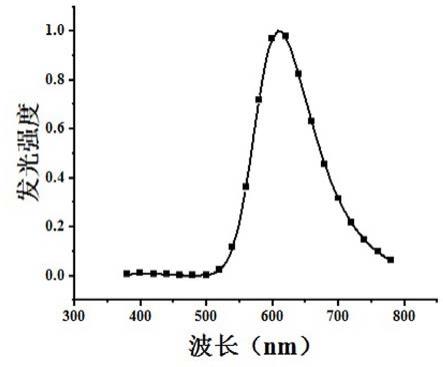
‑
cheng, hao
‑
wu lin, rai
‑
shung liu, and chien
‑
hong cheng, nature photonics, 2018, 12, 235; yasuhiro kondo, kazuki yoshiura, sayuri kitera, hiroki nishi, susumu oda, hajime gotoh, yasuyuki sasada, motoki yanai, and takuji hatakeyama, nature photonics, 2019, 13, 678.)。红光材料由于能隙规则的限制,其非辐射跃迁速率随着发光波长的增加迅速降低,因此高效率、低滚降的红光、深红光tadf材料还十分稀少(见 j. x. chen, w. w. tao, w. c. chen, y. f. xiao, k. wang, c. cao, j. yu, s. li, f. x. geng, c. adachi, c. s. lee, and x. h. zhang, angew. chem. int. ed. engl., 2019, 58, 14660; j. xue, q. liang, r. wang, j. hou, w. li, q. peng, z. shuai, and j. qiao, adv. mater., 2019, 31 , 1808242; y. l. zhang, q. ran, q. wang, y. liu, c. hanisch, s. reineke, j. fan, and l. s. liao, adv. mater., 2019, 31, 1902368.),这限制了高效tadf材料在全彩显示和白
光照明中的应用。
5.因此,现有技术还有待于改进和发展。
技术实现要素:
6.鉴于上述现有技术的不足,本发明的目的在于提供一种具有多发光单元的红光材料及其制备方法和应用,旨在解决现有高效率、低滚降的红光材料较少的问题。
7.本发明的技术方案如下:一种具有多发光单元的红光材料,其中,所述具有多发光单元的红光材料的通式如式(i)所示;式(i);在式(i)中,e基团为给
‑
受体发光单元,所述e基团的通式如式(ii)所示;式(ii);其中,在式(i)中,n为2或3;当n为2时,所述e基团连接在苯环的邻位、对位或间位;当n为3时,所述e基团连接在苯环的均位;在式(ii)中,d基团为给体单元,a基团为受体单元,π环为式(i)中所述e基团所连接的苯环;a基团的分子结构为具体实施方式中a
‑
1~a
‑
7中任何一种。
8.所述具有多发光单元的红光材料,以芳香胺衍生物作为给体单元,以氰基取代的氮杂环作为受体单元,通过两个或三个给
‑
受体单元引入到同一个苯环上,构造具有多发光单元的红光tadf材料,具有较高的摩尔吸光系数以及荧光量子效率,可以实现更高效的红光发射以及制备更高效的电致发光器件。
9.所述的具有多发光单元的红光材料,其中,所述d基团的分子结构为具体实施方式中d
‑
1~d
‑
25中任何一种。给体单元为芳香胺化合物,采用以上给
‑
受体单元的组合可以构建具有扭曲的分子构型,赋予材料tadf特性,提高三线态激子利用率。
10.所述的具有多发光单元的红光材料,其中,在d
‑
1~d
‑
25中,r1为h、f、cl、苯基、咔唑基、二苯胺基、含有1
‑
16个碳的直链或支链烷基、含有1
‑
16个碳的直链或支链烷氧基,x为c(ch3)2、o、s、n
‑
ph。
11.所述的具有多发光单元的红光材料,其中,所述具有多发光单元的红光材料为具体实施方式中化合物1~49中的任意一种。
12.一种如上所述的具有多发光单元的红光材料的制备方法,其中,包括以下步骤:合成中间体;合成最终产物;其中,所述合成中间体的步骤包括以下步骤:将包含给体基团的溴代原料与第一原料偶联反应,生成包含乙炔基团的中间体m1
或m1’;所述第一原料为邻二乙炔基苯、对二乙炔基苯、间二乙炔基苯或均三乙炔基苯;使用高锰酸钾在弱酸性溶剂中氧化中间体m1或m1’的乙炔基基团,生成连二酮中间体m2或m2’;所述合成最终产物的步骤包括以下步骤:中间体m2或m2’与第二原料反应得到终产物;所述第二原料为二氨基马来腈、4,5
‑
二氰基邻苯二胺、6,7
‑
二氰基
‑
2,3
‑
二氨基萘、二氨基修饰的溴代菲或二氨基修饰的溴代苊。
13.所述的具有多发光单元的红光材料的制备方法,其中,所述合成中间体的步骤,具体包括以下步骤:以三乙胺为溶剂,加入包含给体基团的溴代原料与第一原料,加入pd(pph3)2cl2与cui,在氮气条件下加热回流;将反应体系冷却至室温,加入水,采用二氯甲烷萃取,合并有机相,无水硫酸钠干燥,减压除去有机溶剂,进行柱层析分离,得中间体m1或m1’;加入中间体m1或m1’、丙酮、高锰酸钾、水、乙酸,加热回流;趁热过滤除去固体,滤液旋干后使用柱层析分离,得中间体m2或m2’。
14.所述的具有多发光单元的红光材料的制备方法,其中,当所述第二原料为二氨基马来腈、4,5
‑
二氰基邻苯二胺、6,7
‑
二氰基
‑
2,3
‑
二氨基萘时,所述合成最终产物的步骤具体包括以下步骤:在乙酸中,加入中间体m2或m2’与第二原料,氮气条件下加热回流;待反应体系冷却至室温后,加入冰水,搅拌,减压过滤,滤饼经柱层析分离,真空升华得到红色固体;当所述第二原料为二氨基修饰的溴代菲或二氨基修饰的溴代苊时,所述合成最终产物的步骤具体包括以下步骤:在乙酸中,加入中间体m2或m2’与第二原料,氮气条件下加热回流;待反应体系冷却至室温后,加入冰水,搅拌,减压过滤,对滤饼进行干燥处理;加入氰化亚铜的甲基吡咯烷酮溶液,加热回流;加入冰水,搅拌,减压过滤,滤饼经柱层析分离,真空升华得到红色固体。
15.一种如上所述的具有多发光单元的红光材料的应用,其中,将所述具有多发光单元的红光材料用于制备电致发光器件。
16.所述的具有多发光单元的红光材料的应用,其中,所述电致发光器件含有至少一个发光层,所述至少一个发光层中含有至少一种所述具有多发光单元的红光材料;所述具有多发光单元的红光材料占一个所述发光层的质量百分比为0.1~100.0%。
17.所述的具有多发光单元的红光材料的应用,其中,所述有机电致发光器件由下至上依次包括衬底、阳极、空穴传输层、发光层、电子传输层、电子注入层和阴极层。
18.有益效果:本发明的具有多发光单元的红光材料,以芳香胺衍生物作为给体单元,以氰基取代的氮杂环作为受体单元,通过两个或三个给
‑
受体单元引入到同一个苯环上,构造具有多发光单元的红光tadf材料,具有较高的摩尔吸光系数以及荧光量子效率,可以实现更高效的红光发射以及制备更高效的电致发光器件。
附图说明
19.图1为本发明中电致发光器件的结构示意图。
20.图2为本发明中实施例10中掺杂有化合物1的电致发光器件的光谱图。
21.标号说明:1、衬底;2、阳极;3、空穴传输层;4、发光层;5、电子传输层;6、电子注入层;7、阴极层。
具体实施方式
22.本发明提供一种具有多发光单元的红光材料及其制备方法和应用,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,本发明提供了的各种特定的工艺和材料的例子,但是本领域普通技术人员可以意识到其他工艺的应用和/或其他材料的使用。
23.本发明提供一种具有多发光单元的红光材料,所述具有多发光单元的红光材料的通式如式(i)所示;式(i);在式(i)中,e基团为给
‑
受体发光单元,e基团的通式如式(ii)所示;式(ii);其中,在式(i)中,n为2或3;当n为2时,e基团连接在苯环的邻位、对位或间位;当n为3时,e基团连接在苯环的均位;在式(ii)中,d基团为给体单元,a基团为受体单元,π环为式(i)中e基团所连接的苯环。
24.进一步地,d基团的分子结构可以为d
‑
1~d
‑
25中任何一种:25中任何一种:
。
25.在d
‑
1~d
‑
25中,r1可以是h、f、cl、苯基、咔唑基、二苯胺基、含有1
‑
16个碳的直链或支链烷基、含有1
‑
16个碳的直链或支链烷氧基,x为c(ch3)2、o、s、n
‑
ph。
26.进一步地,a基团的分子结构可以为a
‑
1~a
‑
7中任何一种:
。
27.所述受体单元的结构特点为氰基取代的吡嗪及其扩展π
‑
共轭体系衍生物,这种受体单元具有强吸电子能力,适用于红光给
‑
受体结构材料的开发,受体单元刚性强,可以有效的抑制非辐射跃迁,获得较高的荧光量子效率。给体单元为芳香胺化合物,采用以上给
‑
受体单元的组合可以构建具有扭曲的分子构型,赋予材料tadf特性,提高三线态激子利用率。本发明专利中多个给
‑
受体单元的引入可以提高材料的摩尔吸光系数,提高发光效率。
28.在本发明优选实施例方案中,所述具有多发光单元的红光材料可以为化合物1~49中的任意一种。中的任意一种。中的任意一种。
29.。
30.本发明中还提供所述具有多发光单元的红光材料的制备方法,所述具有多发光单元的红光材料的合成通式如式(iii)、式(iv)、式(v)、式(vi)和式(vii)所示。
31.式(iii);式(iv);式(v);式(vi);
式(vii)。
32.具体地,所述具有多发光单元的红光材料的制备方法,包括以下步骤:(1)合成中间体;(2)合成最终产物。
33.其中,所述合成中间体的合成路线如式(iii)所示,具体可以包括以下步骤:(1a)将包含给体基团的溴代原料与第一原料偶联反应,生成包含乙炔基团的中间体m1或m1’,反应条件a为pd(pph3)2cl2,cui,et3n(三乙胺),回流16~24小时。
34.步骤(1a)中,当n为2时,双取代的中间体m1的制备过程中,包含给体基团的溴代原
料与第一原料之间的摩尔比可以为2.2:1或以上;当n为3时,三取代的中间体m1’的制备过程中,包含给体基团的溴代原料与第一原料之间的摩尔比可以为3.3:1或以上。
35.步骤(1a)中, pd(pph3)2cl
2 的用量为第一原料的摩尔量的0.5%~10%,cui的用量为第一原料的摩尔量的10~30%,et3n作为溶剂无严格要求。
36.其中,第一原料可以为二乙炔基苯或三乙炔基苯;所述二乙炔基苯可以为邻二乙炔基苯、间二乙炔基苯或对二乙炔基苯。当第一原料为二乙炔基苯时,得到的中间体对应为m1、m2;当第一原料为均三乙炔基苯时,得到的中间体对应为m1’、m2’。
37.步骤(1a)具体可以为,以三乙胺为溶剂,加入包含给体基团的溴代原料与第一原料,加入pd(pph3)2cl2与cui,在氮气条件下加热回流。回流结束后还可以包括:将反应体系冷却至室温,加入水,采用二氯甲烷萃取,合并有机相,无水硫酸钠干燥,减压除去有机溶剂,进行柱层析分离(洗脱剂可以为二氯甲烷:石油醚体积比1:5),得中间体m1或m1’。
38.(1b)使用高锰酸钾在弱酸性溶剂中氧化中间体m1或m1’的乙炔基基团,生成连二酮中间体m2或m2’,反应条件b为kmno4,haco(乙酸),h2o,acetone(丙酮),回流1~5小时。
39.步骤(1b)中,kmno4的用量为中间体m1或m1
’ꢀ
的摩尔量的4~8倍; haco水溶液浓度为3 mol/l,haco的用量是中间体m1或m1’的的摩尔量2~5倍,丙酮作为溶剂无严格用量限制。
40.步骤(1b)具体可以为,加入中间体m1或m1’、丙酮、高锰酸钾、水、乙酸,加热回流。回流结束后还可以包括:趁热过滤除去固体,滤液旋干后使用柱层析分离(洗脱剂可以为二氯甲烷:石油醚体积比3:2),得中间体m2或m2’。
41.所述合成最终产物的过程,可以包括以下步骤:中间体m2或m2’与第二原料,反应得到终产物。
42.所述第二原料可以为二氨基马来腈、4,5
‑
二氰基邻苯二胺、6,7
‑
二氰基
‑
2,3
‑
二氨基萘、二氨基修饰的溴代菲或二氨基修饰的溴代苊。
43.当所述第二原料为二氨基马来腈、4,5
‑
二氰基邻苯二胺或6,7
‑
二氰基
‑
2,3
‑
二氨基萘时,所述合成最终产物的合成路线如式(iv)或式(v)所示,当采用中间体m2时,合成路线如式(iv)所示;当采用中间体m2’时,合成路线如式(v)所示。所述合成最终产物的过程包括以下步骤:中间体m2或m2’与二氨基马来腈、4,5
‑
二氰基邻苯二胺或6,7
‑
二氰基
‑
2,3
‑
二氨基萘通过扣环反应得到终产物。其中,反应条件c为hoac(乙酸),回流18~24小时。
44.在此步骤中,m2与第二原料的摩尔比例为1:2.2及以下,m2’与第二原料的摩尔比例为1:3.3及以下,hoac作为溶剂无严格要求。
45.进一步地,当所述第二原料为二氨基马来腈、4,5
‑
二氰基邻苯二胺、6,7
‑
二氰基
‑
2,3
‑
二氨基萘时,所述合成最终产物的过程具体包括以下步骤:在乙酸中,加入中间体m2或m2’与第二原料,氮气条件下加热回流;待反应体系冷却至室温后,加入冰水,搅拌,减压过滤,滤饼经柱层析分离(洗脱剂可以为二氯甲烷:石油醚体积比为1:1),真空升华得到红色固体。
46.当所述第二原料为二氨基修饰的溴代菲或二氨基修饰的溴代苊时,所述合成最终产物的合成路线如式(vi)或式(vii)所示,当采用中间体m2时,合成路线如式(vi)所示;当
采用中间体m2’时,合成路线如式(vii)所示。所述合成最终产物的过程包括以下步骤:中间体m2,m2’与第二原料在hoac条件下回流得到氮杂环化合物后,溴原子在cucn(氰化亚铜)的nmp(甲基吡咯烷酮)溶液中被氰基取代得到终产物。其中,反应条件c为hoac(乙酸),回流18~24小时;反应条件d为cucn,nmp回流24~36小时。
47.在此步骤中,hoac作为溶剂,其用量无严格要求。nmp作为溶剂,其用量无严格要求。cucn 的用量为中间体m2,m2’与第二原料在hoac条件下回流得到氮杂环化合物的摩尔量的2~12倍。
48.进一步地,当所述第二原料为二氨基修饰的溴代菲或二氨基修饰的溴代苊时,所述合成最终产物的过程具体包括以下步骤:在乙酸中,加入中间体m2或m2’与第二原料,氮气条件下加热回流18~24小时;待反应体系冷却至室温后,加入冰水,搅拌,减压过滤,对滤饼进行干燥处理;加入氰化亚铜的甲基吡咯烷酮溶液,加热回流24~36小时;加入冰水,搅拌,减压过滤,滤饼经柱层析分离(此处的洗脱剂可以为二氯甲烷:石油醚体积比为1:1),真空升华得到红色固体。
49.其中,干燥处理的方式可以为真空烘箱80℃下烘干,烘干处理时间可以为4~8小时。
50.在本发明优选实施例中,本发明还提供化合物1~49的对应所需的原料,具体见表1所示。
51.表1
化合物编号包含给体基团的溴代原料第一原料第二原料14
‑
溴三苯胺对乙炔基苯二氨基马来腈24
‑
溴三苯胺对乙炔基苯4,5
‑
二氰基邻苯二胺34
‑
溴三苯胺对乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘44
‑
溴三苯胺对乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲54
‑
溴三苯胺对乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲64
‑
溴三苯胺对乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲74
‑
溴三苯胺对乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯84
‑
溴三苯胺间二乙炔基苯二氨基马来腈94
‑
溴三苯胺间二乙炔基苯4,5
‑
二氰基邻苯二胺104
‑
溴三苯胺间二乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘114
‑
溴三苯胺间二乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲124
‑
溴三苯胺间二乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲134
‑
溴三苯胺间二乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲144
‑
溴三苯胺间二乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯159
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯二氨基马来腈169
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯4,5
‑
二氰基邻苯二胺179
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘189
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲199
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲209
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲219
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑对乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯224
‑
溴三苯胺均三乙炔基苯二氨基马来腈234
‑
溴三苯胺均三乙炔基苯4,5
‑
二氰基邻苯二胺
244
‑
溴三苯胺均三乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘254
‑
溴三苯胺均三乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲264
‑
溴三苯胺均三乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲274
‑
溴三苯胺均三乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲284
‑
溴三苯胺均三乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯299
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯二氨基马来腈309
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯4,5
‑
二氰基邻苯二胺319
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘329
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲339
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲349
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲359
‑
(4
‑
溴苯基)
‑
9氢
‑
咔唑均三乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯3610
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯二氨基马来腈3710
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯4,5
‑
二氰基邻苯二胺3810
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘3910
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲4010
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲4110
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲4210
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶对乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯4310
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯二氨基马来腈4410
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯4,5
‑
二氰基邻苯二胺4510
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯2,3
‑
二氰基
‑
6,7
‑
二氨基萘4610
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯3,6
‑
二溴
‑
9,10
‑
二氨基菲4710
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯2,7
‑
二溴
‑
9,10
‑
二氨基菲4810
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲4910
‑
(4
‑
溴苯基)
‑
9,9
‑
二甲基
‑
9,10
‑
二氢吖啶均三乙炔基苯1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯
本发明所提供的具有多发光单元的红光材料,是一类具有多发光单元的橙红光、红光tadf材料,以芳香胺衍生物作为给体单元,以氰基取代的氮杂环作为受体单元,通过两个或三个给
‑
受体单元引入到同一个苯环上,构造具有多发光单元的红光tadf材料,由于多个发光单元的引入,可以提高化合物的摩尔吸光系数与荧光量子产率。因此,该系列化合物具有较高的摩尔吸光系数以及荧光量子效率,可以实现更高效的红光发射以及制备更高效的电致发光器件。
52.本发明中还提供所述具有多发光单元的红光材料的应用,将所述具有多发光单元的红光材料用于制备电致发光器件。所述电致发光器件可以使用现有的蒸镀或者溶液加工的方式制备。所述电致发光器件可用于制备照明光源、信号灯、指示牌或平板显示器。
53.进一步地,所述电致发光器件含有至少一个发光层,至少一个发光层中含有至少一种所述具有多发光单元的红光材料。更进一步地,所述具有多发光单元的红光材料占一个发光层质量的0.1~100.0%,即所述具有多发光单元的红光材料以0.1~100.0%的质量掺杂于主体材料中用于制备发光层。
54.更进一步地,如图1所示,所述有机电致发光器件由下至上依次包括衬底1、阳极2、空穴传输层3、发光层4、电子传输层5、电子注入层6和阴极层7。其中,所述衬底1可以为透明玻璃或其他透明衬底;所述阳极2可以为ito(铟锡氧化物);所述空穴传输层3可以为tapc(4,4'
‑
环己基二[n,n
‑
二(4
‑
甲基苯基)苯胺)等;所述发光层4可以由所述具有多发光单元
的红光材料以0.1~100.0%的质量掺杂于主体材料cbp(4,4'
‑
二(9
‑
咔唑)联苯)中制备得到;所述电子传输层5可以为tmpypb(3,3'
‑
[5'
‑
[3
‑
(3
‑
吡啶基)苯基][1,1':3',1''
‑
三联苯]
‑
3,3''
‑
二基]二吡啶)等;所述电子注入层6可以为lif(氟化锂)等;所述阴极层7可以为金属al(铝)等。
[0055]
其中,所述tapc的分子结构式为,所述cbp的分子结构式为,所述tmpypb的分子结构式为。
[0056]
为进一步说明本发明的具体效果和实施方式,列举如下实施例进行说明,但是,这些实施例不是用来限制本发明。
[0057]
在以下实施例中,化合物的结构是通过质谱和元素分析来确定的,化合物的纯度是通过液相高压色谱仪(hplc)测定的。质谱测试使用的是离子阱型气相色谱
‑
质谱联用仪(mass spectra: ms)
‑
赛默飞世尔(thermo fisher)公司的itq 1100气
‑
质联用仪,元素分析仪使用的是德国elementar公司的vario micro cube。hplc使用安捷伦1200dad高压液相色谱仪测定。
[0058]
实施例1:化合物1的合成化合物1的合成路线如式(viii)所示:式(viii)。
[0059]
(1)化合物b1的合成将化合物4
‑
溴三苯胺 (9.7 g, 30.0 mmol)和对乙炔基苯(1.85 g, 14.6 mmol)加入到250 ml双口瓶中,加入80.0 ml三乙胺溶剂,然后加入pd(pph3)2cl2(631.7 mg, 0.9 mmol)以及cui(171.9 mg, 0.9 mmol),氮气条件下反应体系加热回流24小时。待体系降至室温,将反应体系倒入300 ml水中,使用二氯甲烷萃取三次后,合并有机相,经无水硫酸钠干燥后,减压除去有机溶剂后,进行柱层析分离,洗脱剂为二氯甲烷:石油醚体积比1:5,最
终得到浅黄色固体a1(5.8 g, 产率64.6%)。将化合物a1(5.0 g, 8.2 mmol)加入到250 ml双口瓶中,加入80.0 ml丙酮溶剂,然后加入高锰酸钾(5.2 g , 32.8 mmol),水(6.5 ml),乙酸(2.5 ml),加热回流2小时,趁热过滤除去固体,滤液旋干后使用柱层析分离,洗脱剂为二氯甲烷:石油醚体积比3:2,得到黄色固体b1(4.8 g,产率:87.3%)。
[0060]
(2)化合物1的合成将化合物b1(1.35 g, 2.0 mmol)与二氨基马来腈(0.48 g, 4.4 mmol)溶解到50.0 ml乙酸中,氮气条件下加热回流18小时,待反应体系冷却到室温后,将溶液倒入200.0 ml冰水中,搅拌30分钟后,减压过滤,滤饼经柱层析分离后(洗脱剂:二氯甲烷:石油醚体积比为1:1),真空升华得到红色固体0.98 g(产率:59.8%),经质谱分析、元素分析确认为化合物1。质谱分析确定的分子离子质量为:821.02(计算值为:820.92);理论元素含量(%)c
54
h
32
n3:c,79.01 ; h,3.93 ;n,17.06 ;实测元素含量(%):c,79.01;h,4.08;br,17.08。
[0061]
实施例2:化合物2的合成化合物2的合成路线如式(ix)所示: 式(ix)。
[0062]
化合物2的合成与化合物1的合成相似,使用4,5
‑
二氰基邻苯二胺代替二氨基马来睛,其他条件与化合物1相同。最终经柱层析与升华提纯获得红色固体1.3 g(产率:70.6%),经质谱分析、元素分析确认为化合物2。质谱分析确定的分子离子质量为:920.13(计算值为:920.31);理论元素含量(%)c
62
h
36
n
10
:c,80.85; h,3.94;n,15.21;实测元素含量(%):c,80.91;h,4.08;n,15.28。
[0063]
实施例3:化合物3的合成化合物3的合成路线如式(x)所示:式(x)。
[0064]
化合物3的合成与化合物1的合成相似,使用2,3
‑
二氰基
‑
6,7
‑
二氨基萘代替二氨
基马来睛,其他条件与化合物1相同。最终经柱层析与升华提纯获得红色固体1.2 g(产率:58.8%),经质谱分析、元素分析确认为化合物3。质谱分析确定的分子离子质量为:1020.18(计算值为:1020.34);理论元素含量(%)c
70
h
40
n
10
:c,82.33; h,3.95;n,13.72;实测元素含量(%):c,82.11;h,4.02;n,13.78。
[0065]
实施例4:化合物4的合成化合物4的合成路线如式(xi)所示:式(xi)。
[0066]
化合物c1的合成与化合物1的合成相似。使用3,6
‑
二溴
‑
9,10
‑
二氨基菲代替二氨基马来睛,将化合物b1(1.35 g, 2.0 mmol)与3,6
‑
二溴
‑
9,10
‑
二氨基菲(1.60 g, 4.4 mmol)溶解到50.0 ml乙酸中,氮气条件下加热回流18小时,待反应体系冷却到室温后,将溶液倒入200.0 ml冰水中,搅拌30分钟后,减压过滤,滤饼即为化合物c1。c1经干燥后,加入溶有6倍当量的氰化亚铜(1.07 g, 12.0 mmol)的50.0 ml甲基吡咯烷酮中,回流36 h后,将反应体系倒入300.0 ml冰水中,滤饼经柱层析分离,洗脱剂为二氯甲烷:石油醚体积比为1:1,得到红色固体0.97 g(产率43.2%)。经质谱分析、元素分析确认为化合物4。质谱分析确定的分子离子质量为:1120.15(计算值为:1120.38);理论元素含量(%)c
78
h
44
n
10
:c,83.55; h,3.96;n,12.49;实测元素含量(%):c,83.41;h,4.02;n,12.52。
[0067]
实施例5:化合物5的合成化合物5的合成路线如式(xii)所示:式(xii)。
[0068]
化合物5的合成与化合物4的合成相似,使用2,7
‑
二溴
‑
9,10
‑
二氨基菲代替3,6
‑
二溴
‑
9,10
‑
二氨基菲,其他条件与化合物4相同。最终经柱层析分离后得到红色固体0.81 g(产率35.9%)。经质谱分析、元素分析确认为化合物5。质谱分析确定的分子离子质量为:1120.15(计算值为:1120.38);理论元素含量(%)c
78
h
44
n
10
:c,83.55; h,3.96;n,12.49;实测元素含量(%):c,83.41;h,4.02;n,12.52。
[0069]
实施例6:化合物6的合成化合物6的合成路线如式(xiii)所示:
式(xiii)。
[0070]
化合物6的合成与化合物4的合成相似,使用2,3,6,7
‑
四溴
‑
9,10
‑
二氨基菲代替3,6
‑
二溴
‑
9,10
‑
二氨基菲,氰化亚铜改为12倍当量,其他条件与化合物4相同。最终经柱层析分离后得到红色固体1.3 g(产率53.3%)。经质谱分析、元素分析确认为化合物6。质谱分析确定的分子离子质量为:1220.15(计算值为:1220.36);理论元素含量(%)c
82
h
40
n
14
:c,80.64; h,3.30;n,16.06;实测元素含量(%):c,80.41;h,3.22;n,16.02。
[0071]
实施例7:化合物7的合成化合物7的合成路线如式(xiv)所示:式(xiv)。
[0072]
化合物7的合成与化合物4的合成相似,使用1,2
‑
二胺
‑
5,6
‑
二溴
‑
苊烯代替3,6
‑
二溴
‑
9,10
‑
二氨基菲,其他条件与化合物4相同。最终经柱层析分离后得到红色固体1.0 g(产率47.8%)。经质谱分析、元素分析确认为化合物7。质谱分析确定的分子离子质量为:1068.15(计算值为:1068.34);理论元素含量(%)c
74
h
40
n
10
:c,83.13; h,3.77;n,13.10;实测元素含量(%):c,83.11;h,3.72;n,13.02。
[0073]
实施例8:化合物8的合成化合物8的合成路线如式(xv)和式(xvi)所示:式(xv);
式(xvi)。
[0074]
化合物8的合成与化合物1的合成相似,使用间二乙炔基苯代替对二乙炔基苯,其他条件与化合物1相同。最终得到橙红色固体1.5 g(产率:91.4%)。经质谱分析、元素分析确认为化合物8。质谱分析确定的分子离子质量为:820.15(计算值为:820.28);理论元素含量(%)c
54
h
32
n
10
:c,79.01; h,3.93;n,17.06;实测元素含量(%):c,79.11;h,3.72;n,17.02。
[0075]
实施例9:化合物22的合成化合物22的合成路线如式(xvii)和式(xviii)所示:式(xvii);式(xviii)。
[0076]
化合物22的合成与化合物1的合成相似,使用均三乙炔基苯代替对二乙炔基苯。最终得到红色固体1.7 g(产率:71.3%)。经质谱分析、元素分析确认为化合物22。质谱分析确定的分子离子质量为:1192.10(计算值为:1192.32);理论元素含量(%)c
78
h
45
n
15
:c,78.57; h,3.80;n,17.62;实测元素含量(%):c,78.51;h,3.72;n,17.52。
[0077]
实施例10:发光器件[ito/tapc/cbp:化合物1(10%)/tmpypb/lif/al]在镀有ito阳极的玻璃基片上依次蒸镀空穴传输层、发光层、电子传输层、电子注入层和阴极层,空穴传输层为tapc(厚度为50 nm),发光层为化合物1以10%(质量比)的浓度掺杂于cbp中(厚度为25 nm),电子传输层为tmpypb(厚度为35 nm),电子注入层为lif(厚度
为5 nm),阴极层为金属al阴极(厚度为 100 nm)。在蒸镀的过程中始终保持压强为5*10
‑
4 pa。该器件的开启电压为3.2 v,最大电流效率25.6 cd/a,功率效率29.0 lm/w。该器件发光峰位为612 nm(如图2所示),最大亮度为10590 cd/m2。
[0078]
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1