树脂组合物、复合树脂及其制备与应用的制作方法
1.本发明涉及复合树脂技术领域,特别是涉及一种树脂组合物、复合树脂及其制备与应用。
背景技术:
2.聚酰亚胺是一类高强度、高模量、高耐热和高绝缘性能的高性能材料。已被广泛应用于航空航天、电力电气、微电子等领域。在微电子领域,聚酰亚胺广泛用作挠性印制电路板的绝缘基材。而随着电子工业的飞速发展,电子产品向小型化、高功能化和高安全化方向发展,电子电路朝多层化、高布线密度化的方向发展,由此就对电路板的绝缘基材提出了更高的要求,除了要求其具有较高的玻璃化转变温度及优良的热稳定性之外,对电板基材的热膨胀系数的要求也越来越高。
3.且,为了满足覆铜板对低热膨胀系数的要求,传统技术中常采用低热膨胀系数的聚酰亚胺与热塑性聚酰亚胺进行叠层设置制备绝缘层。然而,不同的功能性聚酰亚胺层的热膨胀系数差异较大,且层间粘接强度不高,容易在覆铜板的焊锡处理过程中、或者在反复折叠过程中分层,造成线电路板的损坏,难以适应柔性电子设备、可穿戴电子设备的应用需求。
4.因此,现有技术仍有待改进。
技术实现要素:
5.基于此,本发明提供了一粘接强度较高且热膨胀系数低的树脂组合物、复合树脂及其制备与应用。
6.本发明的技术方案如下。
7.本发明的一方面提供了一种树脂组合物,包括如下组分:
8.聚酰亚胺a的聚酰胺酸前驱体、聚酰亚胺b的聚酰胺酸前驱体、二维层状纳米填料及添加剂;
9.所述聚酰亚胺a具有式(1)所示结构:
[0010][0011]
其中,所述聚酰亚胺a的各重复单元中的r2各自独立地选自式(1-a)~(1-c)所示结构中的一种:
~c6的烷基,n取1~30的整数;
[0026]
m、p及q表示聚合度,*表示连接位点。
[0027]
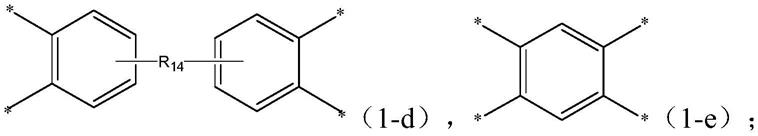
在其中一些实施例中,式(1-d)选自如下任意一种:
[0028][0029]
在其中一些实施例中,所述聚酰亚胺b的各重复单元中的r4各自独立地选自如下所示结构中的一种:
[0030][0031]
其中,ar1、ar2、ar3及ar4分别独立地选自单键或环原子数为6~12的芳基。
[0032]
在其中一些实施例中,所述聚酰亚胺b的各重复单元中的r3各自独立地选自如下所示结构中的一种:
[0033]
和/或
[0034]
所述聚酰亚胺b的各重复单元中的r4各自独立地选自如下所示结构中的一种:
[0035][0036][0037]
在其中一些实施例中,r
11
和r
12
分别独立地选自h、甲基或三氟甲基;和/或
[0038]
r'和r”独立地选自取代或未取代的c1~c6的烷基。
[0039]
在其中一些实施例中,所述聚酰亚胺a的各重复单元中的r2各自独立地选自如下所示结构中的一种:
[0040][0041][0042]
在其中一些实施例中,在所述聚酰亚胺b的聚酰胺酸前驱体中,r4与r5的摩尔量之比为(30~50):(50~70)。
[0043]
在其中一些实施例中,按照质量份数计,所述树脂组合物的组分包括:
[0044][0045]
在其中一些实施例中,所述聚酰亚胺b的聚酰胺酸前驱体与所述二维层状纳米填料的质量比为10:(0.1~5)。
[0046]
本发明的另一方面,提供如上所述的树脂组合物的制备方法,包括如下步骤:
[0047]
将式(i)化合物与式(ii)化合物进行开环聚合反应,得到所述聚酰亚胺a的聚酰胺酸前驱体;
[0048]
将所述二维层状纳米填料、式(iv)化合物、式(v)化合物和有机溶剂混合,得到混合液;
[0049]
将式(iii)化合物与所述混合液进行开环聚合反应,得到所述二维层状纳米填料和所述聚酰亚胺b的聚酰胺酸前驱体的复合物;
[0050]
将所述复合物、所述聚酰亚胺a的聚酰胺酸前驱体和所述添加剂混合,得到所述树脂组合物;
[0051]
其中,式(i)化合物、式(ii)化合物、式(iii)化合物、式(iv)化合物及式(v)化合物的结构如下所示:
[0052]
h2n-r
2-nh2(ii)、
[0053]
h2n-r
4-nh2(iv)、h2n-r
5-nh2(v);
[0054]
r1~r5同权利要求1中r1~r5相同。
[0055]
本发明的又一方面,还提供了一种复合树脂,所述复合树脂采用包括如上所述的树脂组合物的原料制得。
[0056]
本发明的又一方面,提供了一种覆铜板,所述覆铜板包括绝缘基层及设于所述绝缘基层表面的铜箔,所述绝缘基层的制备原料包括如上所述的树脂组合物。
[0057]
进一步地,本发明提供一种印制电路板,所述印制电路板包括如上所述的覆铜板。
[0058]
本发明提供的树脂组合物的组分包括聚酰亚胺a的聚酰胺酸前驱体、聚酰亚胺b的聚酰胺酸前驱体、二维层状纳米填料及添加剂,该树脂组合物脱水固化后得到复合树脂。其中,聚酰亚胺a的聚酰胺酸前驱体转化为具有式(1)所示结构的聚酰亚胺a,其具有特定的刚性,可降低复合树脂的热膨胀系数;聚酰亚胺b的聚酰胺酸前驱体转化具有式(2)所示的聚酰亚胺b,其含有低极性、低表面能的硅氧烷结构,可赋予聚酰亚胺b在复合树脂中向表面迁移的能力,可使聚酰亚胺b出现相分离,进而在复合树脂的表面富集,与此同时,而聚酰亚胺b中同时富有含氮杂环结构,含氮杂环结构可以与金属发生配位作用,以增强复合树脂与铜箔的粘接力,同时硅氧烷结构也进一步增加了复合树脂与铜箔铜的强结合力;且,二维层状纳米填料具有很高的长径比,可限定聚酰亚胺链段的自由移动,降低自由体积,从而可进一步降低复合树脂的热膨胀系数;通过各特定的组分协同,该树脂组合物脱水固化后得到复合树脂具有高粘接强度,且同时兼具低热膨胀系数。
[0059]
进一步地,本发明还提供一种覆铜板,该覆铜板包括绝缘基层及设于绝缘基层表面的铜箔,该绝缘基层的制备原料包括如上所述的树脂组合物。该树脂组合物用于制备覆铜板时,与铜箔的粘接性优异,且能降低覆铜板的热膨胀系数,无需多层聚酰亚胺层叠,只需要采用上述树脂组合物制备一层绝缘基层,即可获得粘接性高且热膨胀系数低的覆铜板。因此,该覆铜板可适应柔性电子设备、可穿戴电子设备的应用需求,从而促进发展高端的集成电路。
具体实施方式
[0060]
为了便于理解本发明,下面将对本发明进行更全面的描述,并给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
[0061]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0062]
在本发明中,同一取代基多次出现时,可独立选自不同基团。如式(2-d)含有多个r'和r”时,则r'和r”可独立选自不同基团。
[0063]
本发明中,“*”表示连接位点。
[0064]
本发明中,“取代或未取代”表示所定义的基团可以被取代,也可以不被取代。当所定义的基团被取代时,应理解为任选被本领域可接受的基团所取代,包括但不限于:c1-30烷基、含有3-20个环原子的杂环基、含有5-20个环原子的芳基、含有5-20个环原子的杂芳基。
[0065]
本发明中,基团中未指明连接位点时,表示基团中任选可连接位点作为连接位点。
[0066]
本发明中,取代基相连的单键贯穿相应的环,表示该取代基可与环的任选位置连接,例如中r与苯环的任一可取代位点相连;进一步地,当环上贯穿有两个取代基的单键时表述两个取代基可与环的任选位置连接,且两个取代基的连接位置不同,例如中,由于苯环上的未被取代的碳原子上均只有一个氢原子,故两个取代基不会与同一苯环上的同一个碳原子相连,换言之,两个取代基与环的连接位点不同。进一步地,当r为h时,此时代表苯环上的氢未被其他取代基取代。
[0067]
本发明中,当两个基团由一连接点连接时,例如中,当r选自单键时,,即代表不需要通过具体的基团连接,直接以单键连接,即为
[0068]
本发明中“烷烃基”指的是烷烃失去一个氢后形成的基团,例如甲烷失去一个氢后形成甲基;“烷烃亚基或亚烷基”指的是烷烃失去两个氢后形成的基团,例如甲烷失去两个氢后形成亚甲基。同理,“含氟烷烃亚基”指的是含氟烷烃失去两个氢后形成的基团。
[0069]
本领域的技术人员基于自身多年在印制线路板领域的研究经验,经过大量创造性的实验探究后,获得本发明的技术方案。
[0070]
本发明实施方式提供了一种树脂组合物,包括如下组分:
[0071]
聚酰亚胺a的聚酰胺酸前驱体、聚酰亚胺b的聚酰胺酸前驱体、二维层状纳米填料及添加剂。
[0072]
聚酰亚胺a具有式(1)所示结构:
[0073][0074]
m代表聚合度,即代表聚酰亚胺a中重复单元的重复次数,换言之,聚酰亚胺a是由m个重复单元连接形成。
[0075]
其中,聚酰亚胺a的各重复单元中的r2各自独立地选自式(1-a)~(1-c)所示结构中的一种:
[0076][0077]r11
和r
12
每次出现时,各自独立地选自h或c1~c5的烷烃基;r
13
每次出现时,分别独立地选自单键、酯基、酰胺基及氧原子;
[0078]
聚酰亚胺a的各重复单元中的r1各自独立地选自式(1-d)~(1-e)所示结构中的一种:
[0079][0080]r14
每次出现时,分别独立地选自单键、c1~c5的烷烃基、羰基及氧原子。
[0081]
可理解,聚酰亚胺a的各重复单元中的r2可相同或不同,r2相同时,即聚酰亚胺a由一种重复单元连接而成,r2不相同时,即聚酰亚胺a由几种r2不同的重复单元连接而成,同理,聚酰亚胺a的各重复单元中的r1可相同或不同,且不同结构的重复单元间没有特定的排列方式,可以是无规。
[0082]
聚酰亚胺b具有式(2)所示结构:
[0083][0084]
其中,聚酰亚胺b的各重复单元中的r3各自独立地选自式(2-a)~(2-c)所示结构中的一种:
[0085][0086]r21
每次出现时,选自单键、羰基、氧原子或碳原子数为1~5、且被氟原子取代的含氟烷烃亚基;
[0087]
r4选自取代或未取代的环原子数为5~10的含氮杂芳基;
[0088]
r5具有式(2-d)所示结构:
[0089][0090]
r'和r”独立地选自取代或未取代的环原子数为6~50的芳基或取代或未取代的c1~c6的烷基,n取1~30的整数;
[0091]
p及q表示聚合度,*表示连接位点。
[0092]
需要说明的是,聚酰亚胺b中的不同结构的重复单元间没有特定的排列方式,可以是无规。
[0093]
上述树脂组合物的组分包括聚酰亚胺a的聚酰胺酸前驱体、聚酰亚胺b的聚酰胺酸前驱体、二维层状纳米填料及添加剂,该树脂组合物脱水固化后得到复合树脂。其中,聚酰亚胺a的聚酰胺酸前驱体转化为具有式(1)所示结构的聚酰亚胺a,其具有特定的刚性,可降低复合树脂的热膨胀系数;聚酰亚胺b的聚酰胺酸前驱体转化具有式(2)所示的聚酰亚胺b,其含有低极性、低表面能的硅氧烷结构,可赋予聚酰亚胺b在复合树脂中向表面迁移的能力,可使聚酰亚胺b出现相分离,进而在复合树脂的表面富集,与此同时,而聚酰胺b中同时富有含氮杂环结构,含氮杂环结构可以与金属发生配位作用,以增强复合树脂与铜箔的粘接力,同时硅氧烷结构也进一步增加了复合树脂与铜箔铜的结合力;且,二维层状纳米填料具有很高的长径比,可限定聚酰亚胺链段的自由移动,降低自由体积,从而可进一步降低复合树脂的热膨胀系数;通过各特定的组分协同,该树脂组合物脱水固化后得到复合树脂具有高粘接强度,且同时兼具低热膨胀系数。
[0094]
在其中一些实施例中,式(1-d)选自如下任意一种:
[0095][0096][0097]
在其中一些实施例中,聚酰亚胺b的各重复单元中的r4各自独立地选自如下所示结构中的一种:
[0098][0099]
其中,ar1、ar2、ar3及ar4分别独立地选自单键或环原子数为6~12的芳基。
[0100]
可理解,当上述结构中的ar1、ar2、ar3及ar4选自单键时,及ar1、ar2、ar3及ar4所在之处为单键;“环原子数”实质所定义的基团中成环的原子的数量。
[0101]
在其中一些实施例中,上述环原子数为6~12的芳基选自:苯、萘及其衍生物。
[0102]
在其中一些实施例中,ar1、ar2、ar3及ar4分别独立地选自单键或苯基。
[0103]
进一步地,聚酰亚胺b的各重复单元中的r4各自独立地选自如下所示结构中的一种:
[0104][0105][0106]
在其中一些实施例中,聚酰亚胺b的各重复单元中的r3各自独立地选自如下所示结构中的一种:
[0107][0108]
在其中一些实施例中,在上述聚酰亚胺b中,以r4与r5的摩尔量总和为基准,r5的含量为40%及以上。
[0109]
进一步地,在上述聚酰亚胺b中,r4与r5的摩尔量之比为(30~50):(50~70)。
[0110]
如此,可进一步提高树脂组合物的粘接性、耐焊锡性及耐热性。
[0111]
可理解,上述聚酰亚胺b中,含r4的重复单元的聚合度p和含r5的重复单元的之比为(30~50):(50~70)。
[0112]
在其中一些实施例中,r'和r”独立地选自取代或未取代的c1~c6的烷基。
[0113]
在其中一些实施例中,r
11
和r
12
分别独立地选自h或甲基。
[0114]
在其中一些实施例中,聚酰亚胺a的各重复单元中的r2各自独立地选自如下所示结构中的一种:
[0115][0116]
在其中一些实施例中,按照质量份数计,上述树脂组合物的组分包括:
[0117][0118]
各特定组分通过特定的配比协调作用,进一步降低树脂组合物的热膨胀系数。
[0119]
进一步地,聚酰亚胺b的聚酰胺酸前驱体与二维层状纳米填料的质量比为10:(0.1~5);优选地,聚酰亚胺b的聚酰胺酸前驱体与二维层状纳米填料的质量比为10:(0.5~5)。
[0120]
在其中一些实施例中,上述二维层状纳米填料可以是阳离子型二维层状材料,主要为层状硅酸盐,包括但不限于:蒙脱土、皂土、贝得土、高岭土、海泡石、凹凸棒石和蛭石中的一种或几种。进一步地,上述二维层状纳米填料中的层间金属阳离子为li
+
、na
+
、k
+
、ca
2+
、mg
2+
或ba
2+
。更优选地,上述二维层状纳米填料为钠基蒙脱土、钠基皂土。
[0121]
上述二维层状纳米填料也可以是阴离子型二维层状材料,具体为二元金属氢氧化物和多元金属氢氧化物中的一种或几种;优选为镁铝水滑石,其层间的阴离子为co3
2-。
[0122]
上述添加剂包括表面偶联剂、润湿流平剂和阻燃剂中的至少一种。
[0123]
上述表面偶联剂、润湿流平剂和阻燃剂可以采用本领域常用的表面偶联剂、润湿流平剂和阻燃剂,此处对表面偶联剂、润湿流平剂和阻燃剂进行举例,包括但不限于如下范围:
[0124]
表面偶联剂可以是硅氧烷表面偶联剂、钛酸酯表面偶联剂,具体的硅烷偶联剂可
以是n-2-(氨基乙基)-3-氨基丙基三甲氧基硅烷、3-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷,3-(2,3-环氧丙氧)丙基三乙氧基硅烷、3-(2,3-环氧丙氧)丙基三甲氧基硅烷、3-巯基丙基三甲氧基硅烷及3-丙烯酰氧丙基三甲氧基硅烷中的至少一种。钛酸酯表面偶联剂可以是钛酸四乙酯、钛酸四丁酯中的一种。
[0125]
润湿流平剂可以是聚醚改性有机硅氧烷、聚酯改性有机硅氧烷。
[0126]
阻燃剂选自含磷阻燃剂,具体可是磷酸酯、亚磷酸酯、次磷酸盐、有机磷、磷杂环化合物、聚合物磷(膦)酸酯、磷腈。优选多聚磷酸、次磷酸盐、磷酸酯及磷腈类阻燃剂。优选地,上述阻燃剂为二乙基次磷酸铝、磷酸苯酯、9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物及六苯氧基环三磷腈。
[0127]
进一步地,以100质量份的聚酰亚胺a的聚酰胺酸前驱体为基准,表面偶联剂的质量份数为0.1份~5份,润湿流平剂的质量份数为0.01~3份,阻燃剂的质量份数为0~10份。
[0128]
本发明的一实施方式还提供如上所述的树脂组合物的制备方法,包括如下步骤s10~s40。
[0129]
步骤s10、将式(i)化合物与式(ii)化合物进行开环聚合反应,得到上述聚酰亚胺a的聚酰胺酸前驱体。
[0130]
式(i)化合物、式(ii)化合物的结构如下所示:
[0131]
h2n-r
2-nh2(ii)。
[0132]
r1和r2与上述r1和r2相同。
[0133]
在其中一些实施例中,式(i)化合物、式(ii)化合物的摩尔量之比为1:9~9:1;进一步优选为1:1。
[0134]
在其中一些实施例中,上述开环聚合反应的温度为0~100℃;进一步优选为1℃~30℃。
[0135]
在其做一些实施例中,上述开环聚合反应的时间为1小时~72小时;进一步优选为5小时~24小时。
[0136]
在其中一些实施例中,步骤s10中,上述开环聚合反应在有机溶剂中进行,进一步地,在极性溶剂中进行;优选地,有机溶剂选自n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、环丁砜、丁内酯、甲酚或环己酮中的任意一种;更优选地,有机溶剂选自n-甲基吡咯烷酮、n,n-二甲基甲酰胺和n,n-二甲基乙酰胺中的至少一种。
[0137]
进一步地,上述有机溶剂的用量以使制得的聚酰亚胺a的聚酰胺酸前驱体的溶液的固含量为5wt%~50wt%为准;进一步以使制得的聚酰亚胺a的聚酰胺酸前驱体的溶液的固含量为15wt%~30wt%为准。
[0138]
步骤s20、将二维层状纳米填料、式(iv)化合物、式(v)化合物和有机溶剂混合,得到混合液。
[0139]
步骤s20中,有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、n-甲基-2-吡咯烷酮、二甲基亚砜、环丁砜、环己酮、间甲酚、苯酚、氯苯酚、甲苯及二甲苯中的至少一种。
[0140]
步骤s30、将式(iii)化合物与上述混合液进行开环聚合反应,得到二维层状纳米填料和聚酰亚胺b的聚酰胺酸前驱体的复合物。
[0141]
在步骤s30中,式(iii)化合物、和上述混合液进行开环聚合反应的过程中,式(iii)、式(iv)及式(v)化合物进行了开环聚合,得到聚酰亚胺b的聚酰胺酸前驱体,且由于开环聚合反应在上述含二维层状纳米填料的混合液中的进行,聚酰亚胺b的聚酰胺酸前驱体会负载在二维层状纳米填料的层间,形成复合物。
[0142]
需要说明的是,步骤s10和步骤s20可以没有特定的先后顺序可以先后进行液可以同时进行;换言之,可以先后进行制备聚酰亚胺a的聚酰胺酸前驱体的步骤s10,也可以先进行制备二维层状纳米填料和聚酰亚胺b的聚酰胺酸前驱体的复合物的步骤s20~30,两者也可以同时进行。
[0143]
其中,式(iii)化合物、式(iv)化合物及式(v)化合物的结构如下所示:
[0144]
h2n-r
4-nh2(iv)、h2n-r
5-nh2(v);
[0145]
r3~r5同上述r3~r5相同。
[0146]
在其中一些实施例中,上述步骤s20中,混合的温度为100℃~180℃,保持0.5h~4h;进一步地,上述步骤s30,开环聚合反应的温度为140~180℃、时间为2h~24h。
[0147]
进一步地,上述步骤s20中的有机溶剂的添加量以使获得的复合物溶液的固含量为5wt%~50wt%为准;优选地,以固含量为10wt%~30wt%为准。
[0148]
进一步地,二维层状纳米填料和聚酰亚胺b的聚酰胺酸前驱体的复合物中,二维层状纳米填料与聚酰亚胺b的聚酰胺酸前驱体的质量比为(0.1~5):10;优选为(0.5~5):10。
[0149]
步骤s40、将上述复合物、上述聚酰亚胺a的聚酰胺酸前驱体和添加剂混合,得到上述树脂组合物。
[0150]
本发明一实施方式,还提供一种复合树脂,该复合树脂采用包括如上所述的树脂组合物的原料制得。
[0151]
该复合树脂粘接强度高,且热膨胀系数低。
[0152]
进一步地,上述复合树脂采用包括如上所述的树脂组合物的原料经脱水固化制得。
[0153]
在其中一些实施例中,上述脱水固化的温度为:300℃~360℃。
[0154]
本发明一实施方式还提供一种覆铜板,该覆铜板包括绝缘基层及设于绝缘基层表面的铜箔,绝缘基层的制备原料包括上所述的树脂组合物。
[0155]
上述树脂组合物用于制备覆铜板时,与铜箔的粘接性优异,且能降低覆铜板的热膨胀系数,无需多层聚酰亚胺层叠,只需要采用上述树脂组合物制备一层绝缘基层,即可获得兼具高粘接性和低热膨胀系数的覆铜板。因此,该覆铜板可适应柔性电子设备、可穿戴电子设备的应用需求,从而促进发展高端的集成电路。
[0156]
进一步地,上述绝缘基层的单面或双面覆有铜箔。
[0157]
在其中一些实施例中,上述覆铜板包括绝缘基层及分别设于绝缘基层两侧的两层铜箔层。
[0158]
进一步地,绝缘基层的厚度为0.5μm~100μm;铜箔层的厚度为2μm~50μm在其中一些实施例中,上述覆铜板的制备方法包括如下步骤s50~s60。
[0159]
步骤s50、将上述树脂组合物的溶液涂布于铜箔的表面,然后于300℃~360℃进行脱水固化,以在铜箔的表面形成绝缘基层,得到中间铜板。
[0160]
可理解,上述铜箔具有两个表面,在上述涂布的步骤中,将上述树脂组合物的溶液涂布于铜箔的其中一个表面。
[0161]
上述树脂组合物在300℃~360℃进行脱水固化,树脂组合物中的聚酰亚胺a的聚酰胺酸前驱体、聚酰亚胺b的聚酰胺酸前驱体转化聚酰亚胺a和聚酰亚胺b,铜箔的表面形成具有上述复合树脂的绝缘基层。
[0162]
进一步地,上述将中间铜板300℃~360℃进行脱水固化的步骤之前,还包括梯度加热的步骤:优选地,以1℃/min~4℃/min的升温速率梯度加热至300℃~360℃。
[0163]
进一步地,上述脱水固化的步骤在惰性气体的保护下进行。
[0164]
步骤s60、将另一铜箔置于上述中间铜板的绝缘基层远离铜箔的一面上,热压,得到覆铜板。
[0165]
在其中一些实施例中,热压的工艺参数为:压力为0.2mpa~50mpa、温度120℃~300℃、时间为0.1min~30min。
[0166]
优选地,热压的工艺参数为:压力为0.2mpa~20mpa、温度150℃~260℃、时间为0.1min~10min。
[0167]
进一步地,本发明一实施方式还提供了一种印制电路板,该印制电路板包括如上所述的覆铜板。
[0168]
上述印制电路板的粘接性能高,热膨胀系数低,且耐热耐耐焊锡。适用于制备耐高温抗老化、集成电路封装、高频高速等高性能印制线路板,从而促进发展高端的集成电路。
[0169]
下面将结合具体的实施例对本发明进行了说明,但本发明并不局限于下述实施例,应当理解,所附权利要求概括了本发明的范围,在本发明构思的引导下本领域的技术人员应意识到,对本发明的各实施例所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。
[0170]
具体实施例
[0171]
实施例1
[0172]
(1)将对苯二胺(5.407g,0.05mol)加入到n,n-二甲基乙酰胺(80g)中,室温25℃搅拌使固体溶解,然后加入联苯二酐(bpda、14.71g,0.05mol),室温25℃搅拌反应14小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0173]
(2)将钠基蒙拓土(1.0g),2,6-二氨基吡啶(0.545g,0.005mol),1,3-双(3-氨基丙基)-1,1,3,3-四甲基二硅氧烷(1.242g,0.005mol)加入到n-甲基吡咯烷酮(20g)和甲苯(16g)中,加热到150℃,保持4h,然后加入均苯二酐(2.182g,0.010mol),加热到180℃反应10h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0174]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(18g),n-2-(氨基乙基)-3-氨基丙基三甲氧基硅烷(0.2g),聚醚改性有机硅氧烷(0.1)和磷酸苯酯(0.8g)在室温搅拌,混合均匀,得到树脂组合物。
[0175]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率2℃/min,梯度加热至300℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.0mpa、温度为180℃下
压合10min,形成双面无胶覆铜板。
[0176]
实施例2
[0177]
(1)将2,2'-二甲基-4,4'-联苯二胺(10.615g,0.05mol),加入到n,n-二甲基甲酰胺(105g)中,室温搅拌使固体溶解。加入均苯二酐(6.544,0.03mol),联苯二酐(5.884g,0.02mol),室温25℃下搅拌反应18小时,形成聚合物固含量18%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0178]
(2)将镁铝水滑石(1.4g),3,5-二氨基吡啶(0.545g,0.005mol),分子量1000的端氨基丙基聚硅氧烷(5.00g,0.005mol)加入到n,n-二甲基甲酰胺(30g)和环己酮(23g)中,加热到160℃,保持3h,然后加入联苯二酐(2.942g,0.01mol),加热到150℃反应10h,形成固含量15%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0179]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(55.6g),聚酰亚胺b的聚酰胺酸前驱体溶液(9g),3-氨基丙基三甲氧基硅烷(0.2g),聚醚改性有机硅氧烷(0.08)和二乙基次磷酸铝(0.4g)在室温搅拌混合均匀,得到树脂组合物。
[0180]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1℃/min,梯度加热至320℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为200℃下压合15min,形成双面无胶覆铜板。
[0181]
实施例3
[0182]
(1)将3,3'-二甲基-4,4'-联苯二胺(10.615g,0.05mol),加入到n-甲基吡咯烷酮(80g)中,室温搅拌使固体溶解,然后加入3,3',4,4'-二苯甲酮四甲酸二酐(16.11g,0.05mol),室温25℃搅拌反应15小时,形成聚合物固含量25%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0183]
(2)将钠基皂土(1.8g),4,4
′‑
二氨基联吡啶(0.931g,0.005mol),分子量2000的端氨基丙基聚硅氧烷(10.00g,0.005mol)加入到环己酮(16g)中,加热到150℃,保持4h,然后加入六氟二酐(4.442g,0.01mol),加热到160℃,反应18h,形成固含量20%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0184]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(40g),聚酰亚胺b的聚酰胺酸前驱体溶液(6g),3-(2,3-环氧丙氧)丙基三甲氧基硅烷(0.5g),聚酯改性有机硅氧烷(0.4g)和9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0185]
(4)将脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至310℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于0.8mpa、温度为190℃下压合15min,形成双面无胶覆铜板。
[0186]
实施例4
[0187]
(1)将联苯二胺(7.370g,0.04mol),4,4
′‑
二氨基二苯醚(2.002g,0.01mol),加入到n,n-二甲基甲酰胺(81g)中,室温搅拌使固体溶解,然后加入均苯二酐(10.906g,0.05mol),室温25℃搅拌反应20小时,形成聚合物固含量20%的聚酰胺酸a溶液。
[0188]
(2)将镁铝水滑石(1.2g),2,5-二(4-氨苯基)吡啶(0.784g,0.003mol),1,3-双(3-氨基丙基)-1,1,3,3-四甲基二硅氧烷(1.738g,0.007mol)加入到n-甲基吡咯烷酮(30g)和
甲苯(21g)中,加热到180℃,保持3h,然后加入二苯醚二酐(3.102g,0.01mol),加热到180℃反应16h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0189]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(19g),钛酸四乙酯(0.2g),聚醚改性有机硅氧烷(0.1)和六苯氧基环三磷腈(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0190]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率2.5℃/min,梯度加热至360℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于2.5mpa、温度为210℃下压合8min,形成双面无胶覆铜板。
[0191]
实施例5
[0192]
(1)将4,4'-二胺苯甲酰替苯胺(6.818g,0.03mol),4,4
′‑
二(4-氨基苯氧基)联苯(0.737g,0.002mol)加入到二甲基亚砜(80g)中,室温搅拌使固体溶解,然后加入均苯二酐(10.91g,0.05mol),室温25℃搅拌反应21小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0193]
(2)将钠基蒙拓土(1.1g),2-(4-氨基苯基)-5-氨基-苯并咪唑(0.45g,0.002mol),分子量1000的端氨基丙基聚硅氧烷(3.00g,0.007mol)加入到n-甲基吡咯烷酮(20g)和甲苯(16g)中,加热到16℃,保持3h,然后加入三苯二醚二酐(4.023g,0.010mol),加热到170℃反应18h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0194]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(11g),3-(丙烯酰氧基)丙基三甲氧基硅烷(0.3g),聚酯改性有机硅氧烷(0.08)和六苯氧基环三磷腈(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0195]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至350℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为220℃下压合15min,形成双面无胶覆铜板。
[0196]
实施例6
[0197]
(1)将4,4'-二氨基苯甲酸苯酯(6.847g,0.03mol),加入到二甲基亚砜(80g)中,室温搅拌使固体溶解,然后加入均苯二酐(6.544g,0.03mol),室温25℃搅拌反应21小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0198]
其中,4,4'-二氨基苯甲酸苯酯的结构如下:
[0199][0200]
(2)将钠基蒙拓土(1.05g),2-(4-氨基苯基)-5-氨基-苯并噁唑(1.126g,0.005mol),分子量1000的端氨基丙基聚硅氧烷(3.00g,0.005mol)加入到n-甲基吡咯烷酮(22g)和甲苯(20g)中,加热到16℃,保持3h,然后加入二苯甲酮二酐(3.222g,0.01mol),加热到170℃反应18h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0201]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(11g),3-(丙烯酰氧基)丙基三甲氧基硅烷(0.4g),聚醚改性有机硅氧烷(0.06)和磷酸苯酯
(0.6g)在室温搅拌,混合均匀,得到树脂组合物。
[0202]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至350℃,形成厚度为20微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为220℃下压合15min,形成双面无胶覆铜板。
[0203]
实施例7
[0204]
(1)将联苯-4,4-二甲酸-二(4-氨基苯酯)(21.223g,0.05mol),加入到n-甲基吡咯烷酮(80g)中,室温搅拌使固体溶解,然后加入联苯二酐(14.71g,0.05mol),室温25℃搅拌反应21小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。联苯-4,4-二甲酸-二(4-氨基苯酯)的结构如下所示:
[0205][0206]
(2)将钠基蒙拓土(1.45g),2-氨基-5-(4-氨苯基)吡啶(0.926g,0.005mol),分子量1000的端氨基丙基聚硅氧烷(5.00g,0.005mol)加入到n-甲基吡咯烷酮(20g)和甲苯(16g)中,加热到16℃,保持3h,然后加入三苯二醚二酐(4.023g,0.010mol),加热到170℃反应18h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0207]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(11g),3-(丙烯酰氧基)丙基三甲氧基硅烷(0.3g),聚酯改性有机硅氧烷(0.08)和六苯氧基环三磷腈(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0208]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至350℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为220℃下压合15min,形成双面无胶覆铜板。
[0209]
实施例8
[0210]
(1)将4,4'-双(4-氨基苯氧基)联苯(18.422g,0.05mol),加入到二甲基乙酰胺(80g)中,室温搅拌使固体溶解,然后加入二苯甲酮二酐(16.11g,0.05mol),室温25℃搅拌反应21小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。
[0211]
(2)将钠基蒙拓土(1.35g),2,5-二氨基咪唑(0.491g,0.005mol),分子量1000的端氨基丙基聚硅氧烷(5.00g,0.005mol)加入到n-甲基吡咯烷酮(25g)和甲苯(20g)中,加热到16℃,保持3h,然后加入三苯二醚二酐(4.023g,0.010mol),加热到170℃反应18h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0212]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(11g),3-(氨丙基)三乙氧基硅烷(0.3g),聚醚改性有机硅氧烷(0.08)和二乙基次磷酸铝(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0213]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至350℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为220℃下压合15min,形成双面无胶覆铜板。
[0214]
实施例9
[0215]
(1)将二(n-4-氨基苯基)-1,1-联苯-4,4-二甲酰胺(21.124g,0.05mol),加入到二甲基亚砜(80g)中,室温搅拌使固体溶解,然后加入二苯甲酮二酐(16.11g,0.05mol),室温25℃搅拌反应21小时,形成聚合物固含量20%的聚酰胺酸a的聚酰胺酸前驱体溶液。二(n-4-氨基苯基)-1,1-联苯-4,4-二甲酰胺的结构如下所示:
[0216][0217]
(2)将钠基蒙拓土(1.50g),2-氨基-5-(4-氨苯基)嘧啶(0.931g,0.005mol),分子量1000的端氨基丙基聚硅氧烷(5.00g,0.005mol)加入到二甲基乙酰胺(20g)和苯(16g)中,加热到16℃,保持3h,然后加入三苯二醚二酐(4.023g,0.010mol),加热到170℃反应18h,形成固含量10%的聚酰亚胺b的聚酰胺酸前驱体溶液。
[0218]
(3)将聚酰胺酸a的聚酰胺酸前驱体溶液(50g),聚酰亚胺b的聚酰胺酸前驱体溶液(11g),3-氨丙基三甲氧基硅烷(0.3g),聚酯改性有机硅氧烷(0.08)和二乙基次磷酸铝(0.5g)在室温搅拌,混合均匀,得到树脂组合物。
[0219]
(4)将树脂组合物均匀涂布于铜箔表面,以升温速率1.5℃/min,梯度加热至350℃,形成厚度为15微米的复合树脂绝缘层,得到无胶单面覆铜板。将制得的无胶单面覆铜板以复合树脂绝缘层的表面与另一铜箔贴合成在一起,在压合机上,于1.5mpa、温度为220℃下压合15min,形成双面无胶覆铜板。
[0220]
对比例1
[0221]
对比例1与实施例1基本相同,不同之处仅在于:步骤(2)中不加钠基蒙拓土,其他条件与步骤同实施例1相同。
[0222]
对比例2
[0223]
对比例1与实施例1基本相同,不同之处仅在于:将实施例1中步骤(1)中的对苯二胺替换成等摩尔质量的如下二氨基萘化合物:
[0224][0225]
其他条件与步骤同实施例1相同。
[0226]
对比例3
[0227]
对比例3与实施例1基本相同,不同之处仅在于:将实施例1步骤(2)中的1,3-二氨丙基六甲基二硅氧烷替换成等摩尔的1,10-二氨基癸烷。
[0228]
其他条件与步骤同实施例1相同。
[0229]
对比例4
[0230]
对比例4与实施例1基本相同,不同之处仅在于:将实施例1步骤(2)中的2,6-二氨基吡啶替换成等摩尔的间苯二胺。
[0231]
其他条件与步骤同实施例1相同。
[0232]
性能测试
[0233]
对实施例和对比例的覆铜板产品的性能进行测试,包括如下步骤:
[0234]
1、玻璃化转变温度的测定:采用动态热机械性能分析仪,rheometricscientific inc,加热速率2℃/min,频率1hz。
[0235]
2、对覆铜板上的复合树脂绝缘层的热膨胀系数(cte)进行测定:采用静态热机械分析仪(tmaq400),测试氛围为氮气,升温速率5℃/min,温度区间为20℃-200℃。
[0236]
3、粘接强度的测定:对于实施例和对比例的覆铜板产品,利用拉力机,测试90
°
拉伸时的剥离强度(n/mm)。
[0237]
4、最高耐焊锡温度的测定:焊锡炉加热到一定温度,实施例和对比例的覆铜板产品固化后,放入焊料浴中以铜箔侧向下的方式漂浮30秒,确认有无外观变化。若无变化,则继续升温。记录覆铜板发生外观变化时,对应焊料浴的最高温度。
[0238]
测试结果如下表1所示:
[0239]
表1
[0240][0241]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0242]
以上实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1