一种合成3-氟-2-氨基异烟腈的方法与流程
1.本发明属于精细化工领域,涉及一种基础医药原材料的合成方法,具体涉及一种合成3-氟-2-氨基异烟腈的方法。
背景技术:
2.吡啶,是含有氮杂原子的六元杂环化合物,与苯相似,具有相同的电子结构,天然存在于煤焦油、页岩油、煤气及石油中。吡啶类化合物作为一种重要的精细化工原料,是目前杂环化合物中开发应用范围最广的品种之一。吡啶类衍生物主要有烷基吡啶、卤代吡啶、氨基吡啶、溴吡啶、甲基吡啶、碘吡啶、氯吡啶、硝基吡啶、羟基吡啶、苄基吡啶、乙基吡啶、氰基吡啶、氟吡啶、二氢吡啶等。其中,吡啶类农药占吡啶系列产品消费总量的50%左右,作为饲料添加剂的吡啶类化合物占比约为30%,医药及其他领域的吡啶类化合物占比约为20%。
3.异烟肼,又名4-吡啶甲酰肼、异烟酸肼,是异烟酸的酰肼,在生物医药领域具有广泛的用途。在吡啶类衍生物合成方面,专利cn201910982605.8公开了3-氟-2-三氟甲基异烟酸的合成方法。3-氟-2-氨基异烟腈作为医药原材料具有一定的应用价值,其现有合成路线有待优化。
技术实现要素:
4.本发明的目的在于优化3-氟-2-氨基异烟腈的合成工艺,为该基础医药原材料的广泛应用提供产业化支撑。
5.为了实现上述目的,本发明提供了一种合成3-氟-2-氨基异烟腈的方法,并给出了合成技术路线。具体地,本发明以2-氯-3-氟吡啶(化合物i)为原料,经锂化(代)反应得到化合物ii,化合物ii经缩醛反应得到化合物iii,化合物iii经c-n偶联反应得到化合物iv,化合物iv经水解反应得到化合物v,化合物v经成肟反应得到化合物vi,化合物vi经酯化反应得到化合物vii,化合物vii经催化脱羧酸反应得到化合物viii,化合物viii经水解脱pmb保护得到目标产物3-氟-2-氨基异烟腈(化合物ix)。
6.由此本领域普通技术人员易于知晓,本发明所涉及的合成方法共八步,进一步的实施方式包括:
7.步骤1),将所述化合物i溶于有机溶剂中,分别依次滴加烷基锂试剂和酰基化试剂,反应完全后,淬灭,得到化合物ii;
8.步骤2),向甲苯-催化剂体系中滴加所述化合物ii和羰基保护试剂,反应液用减液洗涤,得到化合物iii-甲苯的混合溶液;
9.步骤3),向所述化合物iii-甲苯的混合溶液中加入pd催化剂、pd催化剂配体和碱,得到化合物iv;
10.步骤4),将化合物iv加入酸溶液中水解,得到化合物v;
11.步骤5),将所述化合物v溶于有机溶剂中,加入盐酸羟胺和无机碱,反应得到化合
物vi;
12.步骤6),化合物vi酯化反应得到化合物vii;
13.步骤7),将所述化合物vii溶于有机溶剂中,加入三氯化铁、bht,反应得到化合物viii;
14.步骤8),化合物viii酸解后,加入无机碱,得到目标化合物ix。
15.上述步骤1)-8)为制备3-氟-2-氨基异烟腈的优化合成路线,现对本发明所述方法每一步骤涉及的物料、反应条件、操作流程等作出详细的阐述。
16.如上所述步骤1),以2-氯-3-氟吡啶(化合物i)为原料,将化合物i溶于有机溶剂中,控制化合物i-有机溶剂混合体系的温度为-85℃~-80℃;向化合物i-有机溶剂混合体系中缓慢滴加烷基锂试剂,保持反应温度为-85℃~-80℃,搅拌反应1.5h后,控制温度85℃~-80℃;再向反应体系中滴加酰基化试剂,保持反应温度为-85℃~-80℃,搅拌反应完全后,将反应体系加入到酸溶液(优选为稀盐酸溶液)中淬灭,有机相经干燥、浓缩,得到化合物ii。
17.作为步骤1)优选的实施方式,所述有机溶剂可以为四氢呋喃,但不作为步骤1)所述有机溶剂的特别的限定,其它可以实现本发明并能溶解2-氯-3-氟吡啶(化合物i)的有机溶剂均可选用。同样地,所述烷基锂试剂选用甲基锂、乙基锂、丙基锂、异丙基锂、正丁基锂、仲丁基锂、叔丁基锂、戊基锂、己基锂、环己基锂、叔辛基锂、正二十烷基锂、丁基环己基锂中的一种或多种,所述化合物i和烷基锂试剂的摩尔比为1:(1.1~1.3),进一步优选地,烷基锂试剂可以是正丁基锂。所述酰基化试剂选用dmf、甲酸甲酯、甲酸乙酯中的一种或多种,所述化合物i和酰基化试剂的摩尔比为1:(1.2~1.4),进一步优选地,酰基化试剂可以是甲酸乙酯。
18.需要注意的是,酰基化试剂的选用应充分考虑减少杂质的引入,提高目标产物的收率。另外,考虑到化合物ii的稳定性较差,本发明优选采用稀盐酸溶液淬灭反应体系,采用酸性体系减压浓缩的方式,确保化合物ii的稳定性,为下步反应的实施创造条件。
19.如上所述步骤2),向甲苯-催化剂体系中滴加化合物ii和羰基保护试剂。需要特别注意的是,化合物ii的稳定性较差,温度对化合物ii的稳定性有着直接的影响。鉴于此,需要准确控制甲苯-催化剂体系的温度为90℃~92℃,避免化合物ii结构改变及副产物的生成。在搅拌条件下,所述化合物ii和羰基保护试剂的反应温度为100℃~102℃,反应完全后,反应体系的温度控制在35℃~40℃,向反应体系中滴加无机碱溶液洗涤、水洗浓缩得到化合物iii-甲苯的混合溶液。
20.作为步骤2)优选的实施方式,所述羰基保护试剂选用甲醇、乙醇、乙二醇、1.3-丙二醇、硫代甲醇、硫代乙醇、乙二硫醇、1.3-硫代丙二醇、原甲酸三乙酯中的一种或多种,所述化合物ii和羰基保护试剂的摩尔比为1:(1.0~1.2),进一步优选地,所述羰基保护试剂可以是乙二醇。所述甲苯-催化剂体系中的催化剂选用对甲苯磺酸、三氟甲磺酸、浓硫酸、三氟化硼-乙醚、氯化锌、三氟乙酸锌中的一种或多种,所述化合物ii和催化剂的摩尔比为1:(0.05~0.1)。步骤2)中,所述的无机碱为氢氧化钠、氢氧化钾、碳酸钠、叔丁醇钠中的一种或多种,按摩尔比计,所述化合物ii和无机碱的比例为1:(0.1~0.5)。
21.如上所述步骤3),向化合物iii-甲苯的混合溶液中分别添加pd催化剂、pd催化剂配体和强碱,控制温度95℃~100℃,反应完全后,反应液经水洗、干燥、过柱、浓缩、重结晶
得到化合物iv。
22.作为步骤3)优选的实施方式,所述pd催化剂选用醋酸钯、三(二亚苄基丙酮)二钯、[1,1-双(二苯基膦基)二茂铁]二氯化钯中的一种或多种,所述化合物iii和pd催化剂的摩尔比为1:(0.005~0.05)。所述pd催化剂配体选用binap、xphos、sphos中的一种或多种,所述化合物iii和pd催化剂配体的摩尔比为1:(0.015~0.15)。本步骤所用的强碱可以为有机强碱或无机强碱,优选为叔丁醇钠、叔丁醇钾、碳酸钠、碳酸钾、碳酸铯中的一种或多种,所述化合物iii和所述碱的摩尔比为1:(1.5~2.5)。
[0023]
如上所述步骤4),是将化合物iv在特定条件下进行水解反应。本步骤采用稀强酸进行水解,稀强酸水溶液的浓度为1mol/l~10mol/l。优选地,所述强酸选用浓盐酸、甲酸、三氟乙酸中的一种或多种。按摩尔比计,化合物iv与强酸的比例是1:(1.5~2.5),水解反应的温度控制为55℃~60℃,搅拌反应完全后,加入二氯甲烷,滴加无机碱的水溶液,有机相经水洗、干燥、浓缩后得到化合物v。优选地,步骤4)所述的无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钾、碳酸氢钠中的一种或多种,按摩尔比计,化合物iv与无机碱的比例是1:(1.5~2.5)。
[0024]
需要注意的是,化合物iv具有两个反应位点,且反应同时发生,采用步骤4)所述方法可以将副产物的占比控制在10%以下,有效的提高了目标产物的收率。
[0025]
如上所述步骤5),将所述化合物v溶于有机溶剂中,所述有机溶剂优选为二氯甲烷或四氢呋喃,再分别加入盐酸羟胺和无机碱,控制温度20℃~35℃,反应完全后,反应液经干燥浓缩后得到化合物vi。按摩尔比计,化合物v和盐酸羟胺的比例为1:(1.1~2)。所述无机碱选用碳酸钾、氢氧化钠、氢氧化钾、碳酸钠中的一种或多种,按摩尔比计,化合物v和无机碱的比例为1:(1.5~3)。
[0026]
如上所述步骤6),化合物vi酯化反应得到化合物vii。具体地,将化合物vi、乙酸酐溶于甲苯溶剂中,控制反应温度至体系回流(100℃~105℃),搅拌反应完全后,反应液经碱溶液洗涤,水洗、干燥后得到化合物vii-甲苯混合溶液。优选地,所述的化合物vi与乙酸酐的摩尔比为1:(1.0~1.5)。本步骤所述的碱为碳酸钾、氢氧化钠、氢氧化钾、碳酸钠中的一种或多种,按摩尔比计,化合物v和无机碱的比例为1:(1.5~2.0)。
[0027]
需要注意的是,化合物vi具有两个反应位点,且反应同时发生,采用步骤6)所述方法精准控制乙酸酐的用量,避免杂质的生成,有效的提高了目标产物的收率。
[0028]
如上所述步骤7),将所述化合物vii溶于有机溶剂中,加入三氯化铁、bht,反应得到化合物viii。本步骤的有机溶剂选用甲苯,将化合物vii溶于甲苯形成化合物vii-甲苯混合溶液,控制温度100℃~105℃,反应液经无机碱洗涤,水洗、干燥浓缩、柱层析纯化后得到化合物viii。按摩尔比计,化合物vii与三氯化铁的比例为1:(0.02~0.1),化合物vii与bht的比例为1:(0.02~0.1)。
[0029]
如上所述步骤8),化合物viii酸解,所需的酸选用浓盐酸、浓硫酸、三氟乙酸、氢氟酸、甲酸中的一种或多种,化合物viii与强酸的摩尔比为1:(2~5)。本步骤所用无机碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾中的一种或多种,化合物viii与无机碱的摩尔比为1:(1.5~2)。
[0030]
与现有技术相比,本发明所述方法具有如下的有益效果或优点。
[0031]
本发明提供了3-氟-2-氨基异烟腈的新的合成技术路线,其以2-氯-3-氟吡啶(化
合物i)为起始原料,经锂化反应、缩醛反应、c-n偶联反应、水解反应、肟化反应、肟酯化反应、催化脱羧酸反应等步骤得到目标产物。采用本发明方法制备的3-氟-2-氨基异烟腈为浅黄色至土黄色固体,易溶于四氢呋喃,微溶于乙酸乙酯、二氯甲烷。本发明优化了3-氟-2-氨基异烟腈的合成工艺,显著提高了收率和纯度,为3-氟-2-氨基异烟腈作为基础医药原材料的工业化生产创造了条件。
附图说明
[0032]
下面通过附图结合实施例具体描述本发明,其中附图所示内容仅用于对本发明的解释说明,而不构成对本发明任何意义上的限制。
[0033]
图1是本发明实施例化合物ii的lc图谱;
[0034]
图2是本发明实施例化合物iv的gc图谱;
[0035]
图3是本发明实施例化合物v的gc图谱;
[0036]
图4是本发明实施例化合物vi的lc图谱;
[0037]
图5是本发明实施例化合物viii的lc图谱;
[0038]
图6是本发明实施例化合物ix的lc图谱;
[0039]
图7是本发明实施例化合物ix的1hnmr谱图。
具体实施方式
[0040]
以下结合具体实施例,对本发明的具体实施方案做详细的阐述。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围,本发明的保护范围以权利要求为准,包括在此基础上所作出的显而易见的变化或变动等。
[0041]
化合物ii的制备:
[0042][0043]
向3000ml四氢呋喃中加入化合物中i(200g,1.52mol),在-85℃~-80℃下向反应体系中滴加正丁基锂(2mol/l正己烷溶液,900ml,1.82mol),搅拌1.5h后,往体系滴加甲酸乙酯(146g,1.98mol),之后搅拌反应2h,反应结束后,将反应液加入至2mol/l稀盐酸溶液中淬灭至水相ph=2~4,分液,水相用二氯甲烷(500ml
×
2)萃取,有机相合并加入无水硫酸镁干燥,之后有机相浓缩得红棕色油状液体化合物ii:250g,收率95%,lc含量(图1):93.49%。
[0044]
化合物iii的制备:
[0045]
[0046]
向2500ml甲苯溶液中加入对甲苯磺酸(11.54g,0.067mol),在90℃~92℃下,往体系同时滴加化合物ii(208g,1.57mol)和乙二醇(83.21g,1.34mol),滴加完毕后,体系升温至100℃~102℃,搅拌反应4h后,反应体系降温至35℃~40℃,用碳酸钠水溶液洗涤一遍,水洗涤一遍,之后用无水硫酸镁干燥,过滤得到化合物iii-甲苯混合溶液,连投下一步反应。
[0047]
化合物iv的制备:
[0048][0049]
氩气保护下,往化合物iii-甲苯混合溶液中加入4-甲氧基苄胺(215.97g,1.57mol),醋酸钯(0.59g,2.6mmol),binap(4.90g,7.9mmol),叔丁醇钠(252.2g,2.62mol),在95℃~100℃下,搅拌反应4h,反应结束后,反应液水洗一遍,有机相干燥,过柱,过柱液减压浓缩至油状,加入正己烷-甲苯(3ml:5ml)的混合溶液863ml,搅拌下析出固体,过滤得淡黄色化合物iv:350g,收率88.07%,gc含量(图2):96.05%。
[0050]
化合物v的制备:
[0051][0052]
向1104ml 2mol/l稀盐酸溶液中加入化合物iv(350,1.15mol),在55℃~60℃下,搅拌反应7h后,控制温度25℃~30℃,往体系滴加10%碳酸钠溶液,搅拌1h,加入二氯甲烷(1750ml
×
3)萃取,有机相干燥,浓缩至体系为固液状态时,加入正己烷1500ml,搅拌析出固体,搅拌2h后,过滤得到淡黄色固体化合物v:264.83g,收率:85%,gc含量(图3):84.67%。
[0053]
化合物vi的制备:
[0054][0055]
向764.5ml四氢呋喃中,加入化合物v(254.0g,0.95mol),盐酸羟胺(72.61g,1.04mol),乙酸钠(116g,1.41mol),255ml水,室温搅拌反应4h后,将反应液分液,有机相浓缩至油状,加入510ml甲苯,搅拌2h,析出固体,过滤,得到淡黄色固体化合物vi:220g,收率:
84%,lc含量(图4):89.02%。
[0056]
化合物vii的制备:
[0057][0058]
向1970ml甲苯中加入化合物vi(175g,0.72mol)和乙酸酐(73.94g,0.72mol),在100℃~102℃下,搅拌反应2h后,控制温度25℃~30℃,往体系滴加10%碳酸钠溶液,搅拌1h,有机相加入水洗一遍,有机相干燥,得到化合物vii-甲苯甲苯混合液,待投下一步反应。
[0059]
化合物viii的制备:
[0060][0061]
向化合物vii-甲苯混合溶液中,加入三氯化铁(11.59g,0.072mol)和bht(15.76g,0.072mol),在100℃~102℃下,搅拌反应2h后,反应液降温至室温后,硅藻土过滤,滤液加入滴加10%碳酸钠溶液,搅拌1h,有机相加入水洗一遍,有机相干燥,减压浓缩至油状,柱色谱(乙酸乙酯/正己烷)纯化,得到黄色固体:120.07g,收率:65.22%,lc含量(图5):97.87%。
[0062]
化合物ix的制备:
[0063][0064]
向1000ml甲苯中加入化合物viii(100g,0.39mol)和三氟乙酸(221.80g,1.95mol),在80℃~85℃下,搅拌反应3h,将反应液负压浓缩,得到化合物iv的三氟乙酸盐固体,向10%碳酸钠溶液中加入化合物iv三氟乙酸盐,搅拌1h后,过滤,所得固体加入到四氢呋喃溶液中,搅拌溶解,过5μ滤膜,滤液浓缩至固液状,加入甲苯溶剂,搅拌析出固体,得
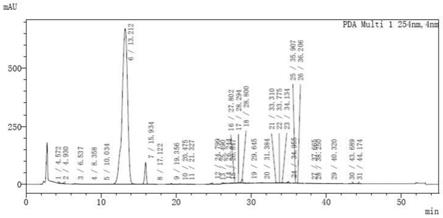
到淡黄色固体化合物ix:44.25g,收率83.02%,lc含量(图6):99.71%,核磁见图7。
[0065]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1