一种胞磷胆碱球形结晶及其制备方法与流程
1.本发明涉及化学工程医药结晶技术领域,具体涉及一种胞磷胆碱球形结晶及其制备方法。
背景技术:
2.胞磷胆碱(cytidinediphosphatecholine,cas:987-78-4);化学中文名:[[(2r, 5r)-5-(4-氨基-2-氧杂环丁-1-基)-3,4-二羟基戊环-2-基]甲氧基-氧化磷酸]2-(三甲胺基) 乙基磷酸酯,其具有如式i所示的化学结构,
[0003][0004]
胞磷胆碱作为磷脂等细胞膜磷脂生物合成的中间体,促进乙酰胆碱等关键神经递质的释放。它是一种注射性神经保护剂,1967年从武田推出,用于治疗缺血性中风后的脑梗死,1968年由西班牙菲尔若(制药)集团研制用于治疗脑血管病,1971年从惠氏制药(现为辉瑞)推出,用于治疗帕金森病。该药物被认为具有多种急性和长期的作用机制,可以限制中风所致的脑损伤,包括通过防止有毒游离脂肪酸的积累来限制梗死范围;通过提供胞苷和胆碱促进脑功能的恢复,这是形成神经细胞膜所必需的两种关键前体,以及促进乙酰胆碱的合成。
[0005]
胞磷胆碱主要用于治疗头部受伤和与脑部手术相关的意识障碍;脑梗塞的情况下急性期意识障碍;中风后偏瘫;胰腺炎胞。辅助治疗帕金森综合征:与抗胆碱药合用对震颤有效;神经性耳聋和耳鸣等。
[0006]
目前,制备较好晶体形态的胞磷胆碱所需结晶工艺要求较高,较难进行工业化大生产。
技术实现要素:
[0007]
有鉴于此,本技术提供一种胞磷胆碱球形结晶的制备方法及一种胞磷胆碱新的晶型,本技术制备方法制备得到球形胞磷胆碱的晶体产品,解决了该产品结晶过程易的成胶问题使产品的流动性优良,堆密度较高,同时提高了原料药的晶型稳定性,防止了胞磷胆碱在储存过程中降解成cmp,增加了药物的稳定性和安全性。
[0008]
为解决以上技术问题,本技术提供的技术方案是一种胞磷胆碱球形结晶的制备方法,包括:
[0009]
胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,加入a溶剂,析出晶体。
[0010]
优选地,所述制备方法具体包括:
[0011]
(1)配制胞磷胆碱-水-a溶剂的混合溶液;
[0012]
(2)将胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,搅拌;
[0013]
(3)加入a溶剂,持续搅拌至出晶,保持搅拌。
[0014]
优选地,所述步骤(1)具体包括:将胞磷胆碱、水和a溶剂混合,得到胞磷胆碱-水-a溶剂的混合溶液。
[0015]
优选地,所述步骤(1)具体包括:
[0016]
将胞磷胆碱和水混合,搅拌,得到胞磷胆碱-水溶液;
[0017]
将胞磷胆碱-水溶液和a溶剂混合,搅拌,得到胞磷胆碱-水-a溶剂的混合溶液。
[0018]
优选地,所述步骤(1)具体包括:在20~30℃条件下,配制胞磷胆碱-水-a溶剂的混合溶液。
[0019]
优选地,所述步骤(1)具体包括:在搅拌条件下,配制胞磷胆碱-水-a溶剂的混合溶液。
[0020]
优选地,所述步骤(1)中搅拌条件为控制搅拌转速为100r/min~250min。
[0021]
优选地,所述步骤(2)具体包括:在30~35℃条件下,将胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,搅拌至出晶。
[0022]
优选地,所述步骤(2)具体包括:控制搅拌转速为100r/min~250min,将胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,搅拌至出晶。
[0023]
优选地,所述步骤(2)具体包括:将胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,搅拌5~60min。
[0024]
优选地,所述步骤(3)具体包括:在30~35℃条件下,加入a溶剂,持续搅拌至出晶,保持搅拌。
[0025]
优选地,所述步骤(3)具体包括:控制搅拌转速为100r/min~250min,加入a 溶剂,持续搅拌至出晶,保持搅拌。
[0026]
优选地,所述步骤(3)具体包括:加入a溶剂,持续搅拌至出晶,保持搅拌转速在100r/min~250r/min,搅拌时间在0.1~5h。
[0027]
优选地,所述制备方法具体包括:
[0028]
(1)在20~30℃,搅拌条件下,配制胞磷胆碱-水-a溶剂的混合溶液;
[0029]
(2)在30~35℃,搅拌转速为100r/min~250min条件下,将胞磷胆碱-水-a溶剂的混合溶剂和架桥剂混合后,搅拌0.1~1h;
[0030]
(3)在30~35℃,搅拌转速为100r/min~250min条件下,加入a溶剂,滴加速率为0.1~5.0wt%/min,持续搅拌至出晶,保持搅拌转速为100r/min~250r/min条件下,搅拌0.1~5h。
[0031]
优选地,所述制备方法还包括:过滤、洗涤、干燥,得到胞磷胆碱球形晶体。
[0032]
优选地,所述制备方法还包括:步骤(4):过滤、洗涤、干燥,得到胞磷胆碱球形晶体。
[0033]
优选地,所述洗涤过程为利用水-a溶剂混合溶液进行润洗。
[0034]
优选地,所述干燥过程燥条件为常压,温度30~40℃,干燥时间12~48h。
[0035]
优选地,所述a溶剂选自乙醇、甲醇、异丙醇、正丁醇、异丁醇、丙酮中的任意一种或多种。
[0036]
优选地,所述架桥剂选自二氧六环、正庚烷、乙酸乙酯、二氯甲烷、正戊烷、环己烷
中的任意一种或多种。
[0037]
优选地,所述胞磷胆碱-水-a溶剂的混合溶液中胞磷胆碱浓度为0.1~0.3g/ml。
[0038]
优选地,所述胞磷胆碱-水-a溶剂的混合溶液中水与a溶剂的质量比为1:1~ 1:5。
[0039]
优选地,架桥剂加入量为所述混合溶剂中胞磷胆碱质量的5~20%。
[0040]
优选地,加入a溶剂过程中a溶剂加入量与所述混合溶剂中a溶剂含量的质量比为0.5:1~2:1。
[0041]
优选地,加入a溶剂过程采用滴加方式加入,滴加速率为0.1~5.0wt%/min。
[0042]
一种胞磷胆碱球形结晶,由上述的制备方法制备得到。
[0043]
优选地,所述球形晶体平均粒径为100~200微米。所述球形晶体颗粒圆润,流动性高。
[0044]
本技术与现有技术相比,其详细说明如下:
[0045]
本发明胞磷胆碱球形结晶制备方法,提高了原料药产品(胞磷胆碱)的后处理能力和后加工性能的同时也简化了单元操作,本发明方法基于球形聚结技术、三元相图设计和优化良溶剂-不良溶剂-架桥剂的溶剂体系、溶剂配比、聚结力和剪切力的平衡关系以及流体力学、调控搅拌力度等晶体工程学的研究,测定了胞磷胆碱在混合溶剂中随温度和组成变化的溶解度和介稳区等热力学数据,建立数学模型,优化最佳的过饱和度曲线,设计最优化的结晶温度、溶剂的滴加速率、总滴加量、结晶时间等参数。基于上述内容,在提供适宜的结晶环境的同时,满足聚结成球的条件要求,最终避免成胶现象,并制备出颗粒圆润,粒度均匀,流动性好,堆密度高的球形产品。
[0046]
本发明方法避免了结晶过程中的油析问题,制备得到的球形粒子产品圆润,粒度均匀,堆密度高且流动性好;且球形药物产品具有较优的填充性,压缩成形性,直接进行压片,省去造粒和造粒后的干燥过程,从而降低工业化成本。
[0047]
本发明所获得得球形结晶,具有高稳定性、高收率、高纯度方面的优点,使得他们可以实现药物制剂通常所需的更严格的药物调节和规格:这些优势一方面有利于用他们制成相应的制剂,比如他们的制剂在制备和并且贮存中具有良好的稳定性与有效性。
附图说明
[0048]
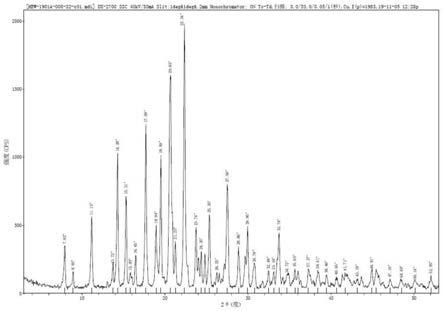
图1为本发明胞磷胆碱球形晶体xrd粉末衍射图谱;图1-1为本发明胞磷胆碱球形晶体xrd寻峰报表图;
[0049]
图2-1为本发明胞磷胆碱球形晶体5倍镜下显微镜照片图;
[0050]
图2-2为本发明胞磷胆碱球形晶体20倍镜下显微镜照片图;
[0051]
图3为对照例2晶体20倍镜下显微镜照片图;
具体实施方式
[0052]
为了使本领域的技术人员更好地理解本发明的技术方案,下面结合具体实施例对本发明作进一步的详细说明。
[0053]
胞磷胆碱为一种多羟基化合物,极易溶于水而不溶于乙醇、甲醇等有机溶剂,因此醇类作为溶析剂,用溶析结晶法为胞磷胆碱结晶的首选。而溶析结晶的关键步骤就是如何控制过饱和度。本发明利用不同检测技术对胞磷胆碱结晶过程进行了研究,结果显示选择
较低的温度和较高的溶析剂浓度有利于诱导晶核形成。对结晶参数进行了优化,结晶温度过高对收率有负面影响,而温度过低则对晶核的形态形成不利;同样,溶析剂流加速度过快会影响结晶的收率和晶型,而太慢的速度会增加企业的生产成本。本发明对结晶产品的粒径分布进行测定,发现采用多种策略对结晶工艺进行控制,获得好的晶型和尺寸分布。
[0054]
专利cn109790197a和文献pharmaceuticalbiotechnology2020,27(2):95~101所述的制备胞磷胆碱的方法中均选择乙醇作为溶析剂,但在析晶过程中极易形成过饱和油状物或黏稠状结晶体,且结晶温度均在45℃以上,故在结晶过程中容易产生降解杂质从而获得的收率相对较低。
[0055]
1982年,日本岐阜药科大学的kawashima在一步过程中完成了析出结晶和聚结成球形颗粒的过程,大大简化了传统制剂工艺,这种在液相中一步完成结晶和聚结成球的过程,就是由kawashima首次提出的“球形结晶”的概念。这一过程操作简单,对设备并无特殊要求,对很多种物系均适用,因而在其提出之后,就引起了众多领域科研人员的研究兴趣,人们对不同物系及不同条件下的球形聚结研究之后,归纳了球形结晶常用的方法,并对其机理也做了一定的推测。
[0056]
若直接将胞磷胆碱溶解于单一溶剂中,通过冷却结晶或溶析结晶的方法,则并不能控制晶体的成球过程。这是由于此类方法无法满足实现聚结力与剪切力的匹配,晶体无法聚结成球。
[0057]
本发明方法使用的特定的溶剂、溶剂配比、晶种加入量等是综合考虑了晶体的成核生长、聚结与破碎、产品性能、工艺的可操作性、生产效率和经济效益的优化方案成本。
[0058]
因此,目前胞磷胆碱产品结晶主要的问题为:在制备过程中易油析,得到最终产品形态差,堆密度低,稳定性较差,要制备较好晶体形态的胞磷胆碱所需结晶工艺要求较高,较难进行工业化大生产。
[0059]
本发明提供了一种避免油析、所得产品形态规整、堆密度高的胞磷胆碱球形结晶制备方法。
[0060]
实施例1:结晶体系为水-甲醇-正庚烷
[0061]
一种胞磷胆碱球形结晶的制备方法
[0062]
步骤1、在20℃下,将胞磷胆碱100g、水100g加入2l的反应釜内搅拌溶解,直至搅拌溶液澄清,得到胞磷胆碱-水溶液。
[0063]
步骤2、将温度升至30℃并保持温度在30℃~35℃,转速调至180r/min,将甲醇300g 加入步骤1的2l反应釜中搅拌10min,加入正庚烷10g,,转速保持180r/min,搅拌15min 后体系浑浊析出大量固体。
[0064]
步骤3,保持釜内温度30℃~35℃,转速调至150r/min,滴加甲醇600g至步骤2反应液中,搅拌下滴加时间为2~3h,滴加速率为0.1~5.0wt%/min(每分钟滴加0.1~5.0wt%的甲醇),滴加完毕后,转速保持150r/min,继续搅拌1h~2h。
[0065]
步骤4,过滤、洗涤、干燥,得到胞磷胆碱球形晶体92.8g;
[0066]
所述洗涤过程为利用水-a溶剂混合溶液进行润洗,水-a溶剂。
[0067]
所述干燥过程燥条件为常压,温度30~40℃,干燥时间12~48h。
[0068]
产品的xrd图谱见图1;球形晶体显微镜照片图见图2-1(放大倍数:5倍)、图 2-2(放大倍数:20倍)
[0069]
本专利制备得到的产品明显呈球形;产品平均粒度在100~150微米之间。
[0070]
实施例2:结晶体系为水-乙醇-正庚烷
[0071]
一种胞磷胆碱球形结晶的制备方法
[0072]
步骤1、在20℃下,将胞磷胆碱100g、水100g加入1l的反应瓶内搅拌溶解,直至搅拌溶液澄清,为胞磷胆碱-水溶液。
[0073]
步骤2、将温度升至30℃并保持温度在30℃~35℃,转速调至180r/min,缓慢将乙醇400g加入步骤1的2l反应釜中搅拌10min,加入正庚烷10g,转速保持至180r/min,搅拌15min后体系浑浊析出少量固体并出现少量油析现象。
[0074]
步骤3,保持釜内温度30℃~35℃,转速调至150r/min缓慢加入乙醇900g至步骤2 反应液中,搅拌下滴加时间为2~3h,滴加速率为0.1~5.0wt%/min(每分钟滴加0.1~ 5.0wt%的甲醇),滴加完毕后,转速保持150r/min,保持搅拌1h~2h,使晶体老化聚结成紧实球体。
[0075]
步骤4,过滤、洗涤、干燥,得到胞磷胆碱球形晶体93.1g;
[0076]
所述洗涤过程为利用水-a溶剂混合溶液进行润洗,水-a溶剂。
[0077]
所述干燥过程燥条件为常压,温度30~40℃,干燥时间12~48h。
[0078]
实施例3:结晶体系为水-异丙醇-正庚烷
[0079]
一种胞磷胆碱球形结晶的制备方法
[0080]
步骤1、在20℃下,将胞磷胆碱100g、水100g加入2l的反应釜内搅拌溶解,直至搅拌溶液澄清,为胞磷胆碱-水溶液。
[0081]
步骤2、将温度升至30℃并保持温度在30℃~35℃,转速调至180r/min,缓慢将异丙醇390g加入步骤1的2l反应釜中搅拌10min,加入正庚烷10g,转速保持至180r/min,搅拌15min后体系浑浊析出大量固体。
[0082]
步骤3,保持釜内温度30℃~35℃,转速调至150r/min,滴加异丙醇780g至步骤2 反应液中,搅拌下滴加时间为2~3h,滴加速率为0.1~5.0wt%/min(每分钟滴加0.1~ 5.0wt%的甲醇),滴加完毕后,保持搅拌1h~2h,使晶体聚结成紧实球体。
[0083]
步骤4,过滤、洗涤、干燥,得到胞磷胆碱球形晶体92.4g。
[0084]
实施例4:结晶体系水-甲醇-1,4二氧六环
[0085]
一种胞磷胆碱球形结晶的制备方法
[0086]
步骤1、在20℃下,将胞磷胆碱100g、水100g加入2l的反应釜内搅拌溶解,直至搅拌溶液澄清,为胞磷胆碱-水溶液。
[0087]
步骤2、将温度升至30℃并保持温度在30℃~35℃,转速调至180r/min,缓慢将甲醇300g加入步骤1的2l反应釜中搅拌10min,加入1,4-二氧六环10g,转速保持至 180r/min,搅拌15min后体系浑浊析出大量固体。
[0088]
步骤3,保持釜内温度30℃~35℃,转速调至150r/min,滴加甲醇至步骤2反应中,搅拌下滴加时间为2~3h,滴加速率为0.1~5.0wt%/min(每分钟滴加0.1~5.0wt%的甲醇),滴加完毕后,保持搅拌1h~2h,使晶体聚结成紧实球体。
[0089]
步骤4,过滤、洗涤、干燥,得到胞磷胆碱球形晶体93.2g;
[0090]
所述洗涤过程为利用水-a溶剂混合溶液进行润洗,水-a溶剂。
[0091]
所述干燥过程燥条件为常压,温度30~40℃,干燥时间12~48h。
[0092]
实施例5:工业化生产
[0093]
一种胞磷胆碱球形结晶的制备方法
[0094]
步骤1、在20℃下,将胞磷胆碱20kg、水20kg加入2000l的反应釜内搅拌溶解,直至搅拌溶液澄清,为胞磷胆碱-水溶液。
[0095]
步骤2、将温度升至30℃并保持温度在30℃~35℃,加入正庚烷2kg,搅拌30min 后体系浑浊析出固体。,缓慢将甲醇60kg加入步骤1的2000l反应釜中搅拌10min,搅拌15min~30min后体系浑浊析出大量固体。
[0096]
步骤3,保持釜内温度30℃,滴加甲醇120kg至步骤2反应液中,搅拌下滴加时间为2~3h,滴加速率为0.1~5.0wt%/min(每分钟滴加0.1~5.0wt%的甲醇),滴加完毕后,保持搅拌1h~2h,使晶体老化,聚结成紧实球体。
[0097]
步骤4,将温度降至25℃后过滤、洗涤、干燥,得到胞磷胆碱球形晶体18.9kg;
[0098]
所述洗涤过程为利用水-a溶剂混合溶液进行润洗,水-a溶剂。
[0099]
所述干燥过程燥条件为常压,温度30~40℃,干燥时间12~48h。
[0100]
对照例1
[0101]
参照专利cn109790197a制备胞磷胆碱晶体
[0102]
取胞磷胆碱127g、水260g加入5l的反应瓶内升温至45℃搅拌至溶清,溶清后升温至60℃并在此温度下缓慢加入乙醇256g,滴加时间为搅拌10min后,维持温度在 60℃下滴加乙醇512g,滴加时间在2h~2.5h。
[0103]
维持温度不变,取0.51g胞磷胆碱晶种加入体系搅拌30min,体系逐渐浑浊,析出大量固体,维持转速和温度继续用2h滴加512g乙醇,滴加完毕后,体系析出大量固体并且出现少量油析现象,将结晶体系由60℃缓慢冷却到25℃后,体系出现大量油析现象,充分搅拌10小时后,将结晶反应液过滤得到产品用80%乙醇进行清洗,减压条件下、50℃下干燥3小时,得到86.36g的胞磷胆碱产品。
[0104]
对照例2
[0105]
参照专利文献日本特公昭51-32630号公报制备胞磷胆碱晶体
[0106]
步骤1、在20℃下,将市售胞磷胆碱127g、水127g加入2l的反应釜内搅拌溶解,直至搅拌溶液澄清配制为胞磷胆碱-水溶液。将温度升至30℃并保持温度在 30℃~35℃,转速调至180r/min,缓慢将甲醇381g加入胞磷胆碱-水溶液的2l反应瓶中搅拌10min,加入胞磷胆碱晶种1.27g,转速保持至180r/min,搅拌5min后体系浑浊析出大量固体。保持釜内温度30℃~35℃,转速调至150r/min,滴加甲醇762g 至上述结晶体系中,搅拌下滴加时间为2~3h,加完毕后,保持搅拌1h~2h,使晶体老化。停止搅拌降至25℃后,过滤、洗涤、干燥,得到胞磷胆碱棒状晶体100.5g,产品显微镜照片图见图3(放大倍数:20倍)。
[0107]
效果例1
[0108]
对实施例1制备得到的胞磷胆碱球形结晶按照药典方法进行稳定性实验考察
[0109]
考察条件和结果如表1所示。
[0110]
表1
[0111][0112]
表1中含量为质量百分含量;表1中杂质1为胞苷酸,结构为:
[0113][0114]
杂质2为尿苷,结构为:
[0115][0116]
由表1可知本发明得到的胞磷胆碱球形结晶稳定性佳。
[0117]
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1