一种小鼠肺微血管内皮细胞永生化以获得胞外囊泡的方法
1.本发明涉及细胞培养技术领域,具体是一种小鼠肺微血管内皮细胞永生化以获得胞外囊泡的方法。
背景技术:
2.肺微血管内皮细胞(pmecs)是肺泡毛细血管屏障的重要组成部分,参与气体交换。它们在肺内稳态和炎症反应中发挥重要作用。鉴于pmecs的重要作用,体外培养pmecs对于阐明肺部疾病的分子和细胞机制至关重要。小鼠肺微血管内皮细胞(pmpmecs)的研究受到细胞培养的限制。目前已经发表的分离pmpmecs最常见的方法包括:肺组织的酶解和pmpmecs对cd31或cd102磁珠免疫结合法。但是,pmpmecs的阳性磁珠选择会导致细胞早期衰老,增殖能力低。此外,磁珠的非特异性结合通常会导致肺组织中其他细胞(如成纤维细胞、平滑肌细胞、上皮细胞)的污染。
3.pmecs分泌的细胞外囊泡(pmec-evs)直接分泌到血流中,并能与循环白细胞相互作用,调节局部免疫反应。越来越多的研究表明,pmec-evs与炎症性肺部疾病的病理生理有关,而pmec-evs介导肺部疾病的具体作用机制研究尚处于探索阶段。由于pmpmecs分泌ev的能力有限,且缺乏可靠的pmpmecs细胞系。许多研究者在研究中使用脐静脉内皮细胞来源的囊泡而不是pmec-evs。然而,囊泡具有高度的异质性,不同来源的囊泡特性差异很大。因此,使用其他内皮细胞来源的囊泡来探索pmecs-evs在肺部疾病中的作用可能会导致研究偏倚。
技术实现要素:
4.本发明的目的在于提供一种小鼠肺微血管内皮细胞永生化以获得胞外囊泡的方法,解决了上述技术问题,避免了传统磁珠阳性选择提取细胞的损害,极大改善内皮细胞的损耗和细胞老化问题;通过转染慢病毒至原代细胞,构建永生化细胞,永生化细胞分泌囊泡保留了原代细胞分泌囊泡的特性,极大降低囊泡提取效率和成本。
5.本发明的目的可以通过以下技术方案实现:
6.一种小鼠肺微血管内皮细胞永生化以获得胞外囊泡的方法,包括以下步骤:
7.s1、小鼠肺微血管内皮原代细胞分离
8.1)小鼠肺缘单细胞悬液的制备
9.分离小鼠肺组织,组织碎化并制备单细胞悬液;
10.2)小鼠肺缘单细胞培养
11.将单细胞悬液加入完全培养基培养至细胞融合度90%以上;
12.3)小鼠肺微血管内皮细胞的分离纯化
13.对重悬后的单细胞进行阴性选择:利用抗小鼠cd45磁珠、抗小鼠cd90.2磁珠和抗小鼠cd326磁珠分别去除淋巴细胞、成纤维细胞和上皮细胞;
14.s2、内皮细胞永生化
15.s2.1将500ul小鼠肺微血管内皮原代细胞悬液种于胶原包被的24孔板中(3
×
104个细胞/孔),培养24h。
16.s2.2将含有gfp的慢病毒(1x 107iu/ml)在37℃水浴中解冻,解冻后立即将其从水浴中移出。然后加上30、60、90、120和150μl慢病毒原液(含5μlviralentry),建立不同moi(moi=10,20,30,40,50)。使用mpmecs培养基将体积调整为500μl。
17.s2.324孔板4000转/分,离心30分钟,以增加转导效率,然后在培养箱中(37℃,5%co2)孵育72h。
18.s2.4倒置荧光显微镜拍摄不同的mois下细胞36、48、60、72h的照片,评估不同moi、时间下病毒转导率。
19.s2.5置于37℃水浴中解冻慢病毒(lenti-sv40,lenti-sv40t,lenti-sv40 t(puro),lenti-rasv12,lenti-htert,lenti-hpv-16e6/e7(puro)),解冻后立即从水浴中移除。然后,在含有5μl viralentry的mpmecs培养基中加入适量的慢病毒颗粒。
20.s2.6将s2.5中含有viralentry和慢病毒的mpmecs培养基在培养箱中共孵育48h后,更换mpmecs培养基。
21.s2.7当细胞90-95%汇合时,消化细胞。将第一次转导的pmpmecs在胶原包被的24孔板上重新播种,每2天更换培养基,重复s2.5-s2.6重复慢病毒转导,并监测细胞生长速率和形态,以选择永生的候选细胞。
22.s3、小鼠肺微血管内皮细胞囊泡提取。
23.进一步地,所述小鼠肺缘单细胞悬液的制备的具体操作步骤:
24.于无菌培养皿取出小鼠双肺,剥离脏层胸膜后剪去两侧肺缘,置于dmem-f12基础培养基中,漂洗3次后移入5ml规格的离心管中,加入适量i型胶原酶工作液。
25.剪碎组织,补齐胶原酶工作液至10ml,将混合液转入c管中,并置于组织研磨仪处理。
26.处理后将c管置于恒温水平摇床中震荡30min,再置于组织研磨仪处理,得到混悬液通过70um尼龙滤网过滤后离心,dmem-f12基础培养基清洗3次后加入红细胞裂解液充分裂解、终止、离心后计算活细胞数量,得到肺缘单细胞悬液。
27.进一步地,所述小鼠肺缘单细胞培养步骤为:
28.将单细胞悬液加入完全培养基后重悬、调整细胞密度,置于t-75培养瓶中培养24小时后弃去上清,重新补充完全培养基,每隔两天换液、观察,直至细胞融合度至90%以上。
29.进一步地,所述小鼠肺微血管内皮细胞的分离纯化步骤包括:
30.将肺缘单细胞消化重悬为单细胞悬液后,利用抗小鼠cd45磁珠、抗小鼠cd90.2磁珠和抗小鼠cd326磁珠分别去除淋巴细胞、成纤维细胞和上皮细胞。
31.将阴性选择剩余的细胞重新置于培养皿中培养,2天更换一次培养基并通过光镜观察,利用记号笔圈出内皮细胞集落边缘后,再利用克隆环选出内皮细胞集落。
32.进一步地,所述步骤1)中选用出生7-14天乳鼠。
33.进一步地,所述i型胶原酶工作液为i型胶原酶溶解于含有血清的完全培养基中。
34.进一步地,所述红细胞裂解液加入10倍体积完全培养基终止。
35.进一步地,所述完全培养基含有92mg/l d-valine,100iu/ml肝素,1%ecgs。
36.进一步地,所述s2中助转染试剂选用viralentry。
37.所述慢病毒包括lenti-sv40,lenti-sv40t,lenti-sv40 t(puro),lenti-rasv12,lenti-htert,lenti-hpv-16e6/e7(puro)。
38.进一步地,所述囊泡的提取采用超速离心法。所述内皮细胞集落通过流式细胞术、免疫荧光的手段检测。
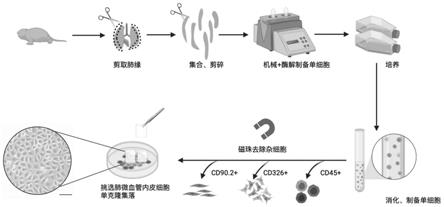
39.本发明的有益效果:
40.1、本发明通过磁珠阴性选择和圈选手段,避免了传统磁珠阳性选择提取细胞的损害,极大改善内皮细胞的损耗和细胞老化问题;
41.2、本发明通过转染慢病毒至原代细胞,构建永生化细胞,永生化细胞分泌囊泡保留了原代细胞分泌囊泡的特性,极大降低囊泡提取效率和成本。
附图说明
42.下面结合附图对本发明作进一步的说明。
43.图1是本发明小鼠肺微血管内皮细胞提取的示意图;
44.图2是本发明小鼠肺微血管内皮细胞光镜图;
45.图3是本发明小鼠肺微血管内皮细胞表面分子鉴定图;
46.图4是本发明小鼠肺微血管内皮细胞成管能力和吞噬能力鉴定图;
47.图5是本发明小鼠肺微血管内皮细胞永生化构建流程图;
48.图6是本发明本发明小鼠肺微血管内皮细胞转染不同慢病毒后形态图;
49.图7是本发明永生化肺微血管内皮细胞表面分子鉴定图;
50.图8是本发明永生化肺微血管内皮细胞成管能力和吞噬能力鉴定图;
51.图9是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)基因表达和增殖比较图;
52.图10是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)渗透性功能比较图;
53.图11是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)炎症刺激反应比较图;
54.图12是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)分泌的囊泡形态比较图;
55.图13是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)分泌的囊泡粒径分布比较图;
56.图14是本发明原代肺微血管内皮细胞(pmpmecs)和永生化肺微血管内皮细胞(impmecs)分泌的囊泡粒径分布比较图。
具体实施方式
57.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
58.s1、小鼠肺微血管内皮原代细胞分离
59.s1.1小鼠肺缘单细胞悬液的制备
60.处死小鼠后于无菌培养皿取出小鼠双肺,剥离脏层胸膜后剪去两侧肺缘,置于dmem-f12基础培养基中,漂洗3次后移入5ml规格的离心管(eppendorf管)中,加入适量i型胶原酶工作液(i型胶原酶溶解于含有血清的完全培养基中);
61.用眼科剪充分剪碎组织后,补齐胶原酶工作液至10ml后,将混合液转入美天妮gentlemacs c管(简称“c管”)中,然后置于美天妮gentlemacs组织研磨仪并运行程序(m-lung-01-02)。
62.程序结束后将c管直接置于恒温水平摇床(37度,200rpm)中震荡30min,然后置于美天妮gentlemacs组织研磨仪运行程序。所得混悬液通过70um尼龙滤网过滤后离心,dmem-f12基础培养基清洗3次后加入红细胞裂解液充分裂解、终止、离心后计算活细胞数量,得到肺缘单细胞悬液。
63.s1.2小鼠肺缘单细胞培养:将s1.1中的到的单细胞悬液加入完全培养基后重悬、调整细胞密度,置于t-75培养瓶中培养24小时后弃去上清,重新补充新鲜完全培养基。每隔两天换液、观察,直至细胞融合度至90%以上。
64.完全培养基是在基础培养基中加入92mg/l d-valine、100iu/ml肝素和1%ecgs。d-valine用于抑制成纤维细胞的过度繁殖;肝素保护内皮细胞糖萼,有助于维持内皮细胞正常结构和功能;ecgs含有内皮细胞生长繁殖需要的各类微量元素和矿物质,能够最大程度的保持内皮细胞的活力和增殖。
65.s1.3小鼠肺微血管内皮细胞的分离纯化:将肺缘单细胞消化重悬为单细胞悬液后,对重悬后的单细胞进行阴性选择:利用抗小鼠cd45磁珠、抗小鼠cd90.2磁珠和抗小鼠cd326磁珠分别去除淋巴细胞、成纤维细胞和上皮细胞。
66.将阴性选择剩余的细胞重新置于培养皿中培养,2天更换一次培养基并通过光镜观察。融合后的肺微血管内皮细胞呈鹅卵石样,利用记号笔圈出内皮细胞集落边缘后,利用克隆环选出内皮细胞集落(内皮原代细胞),如图1所示。所得细胞可通过流式细胞术、免疫荧光等手段检测,结果如图2至图4所示。
67.s2、内皮细胞永生化
68.s2.1将500ul小鼠肺微血管内皮原代细胞悬液种于胶原包被的24孔板中(3
×
104个细胞/孔),培养24h。
69.s2.2将含有gfp的慢病毒(1x107iu/ml)在37℃水浴中解冻,解冻后立即将其从水浴中移出。然后加上30、60、90、120和150μl慢病毒原液(含5μlviralentry),建立不同moi(multiplicity of infection)(moi=10,20,30,40,50)。使用mpmecs培养基将体积调整为500μl。
70.传统的慢病毒助转剂polybrene转染效率低,对原代细胞毒性大,而viralentry利用最新聚合技术在提升转染效率的同时,最大程度的保护原代细胞,确保转染时不会引起原代细胞的死亡和后续改变。
71.s2.324孔板4000转/分,离心30分钟,以增加转导效率,然后在培养箱中(37℃,5%co2)孵育72h。
72.s2.4倒置荧光显微镜拍摄不同的mois下细胞36、48、60、72h的照片,评估不同moi、时间下病毒转导率。
73.s2.5置于37℃水浴中解冻慢病毒(lenti-sv40,lenti-sv40t,lenti-sv40 t(puro),lenti-rasv12,lenti-htert,lenti-hpv-16e6/e7(puro)),解冻后立即从水浴中移除。然后,在含有5μl viralentry的mpmecs培养基中加入适量的慢病毒颗粒,本发明中moi优选30。
74.s2.6将s2.5中含有viralentry和慢病毒的mpmecs培养基在培养箱中共孵育48h后,更换新鲜mpmecs培养基。
75.s2.7当细胞90-95%汇合时,消化细胞。将第一次转导的pmpmecs在胶原包被的24孔板上重新播种,每2天更换培养基,重复s2.5-s2.6重复慢病毒转导,并监测细胞生长速率和形态,以选择永生的候选细胞,构建流程如图5所示。
76.图6为小鼠肺微血管内皮细胞转染不同慢病毒后形态图;图7至图9为构建成功的永生化细胞的形态和功能。
77.s3、小鼠肺微血管内皮细胞囊泡提取
78.超速离心法提取囊泡并鉴定囊泡性质。原代细胞和永生化细胞分泌的囊泡特征如图12至图14所示。
79.在本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
80.以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1