螺环嘧啶酮衍生物、其制备方法、药物组合物和用途与流程
1.本发明涉及新的嘧啶酮化合物、其制备方法及包含该化合物的药物组合物以及它们在治疗由lp-pla2介导的疾病中的用途。
背景技术:
2.脂蛋白相关磷脂酶a2(lp-pla2)是参与脂蛋白脂质或磷脂的水解的磷脂酶a2酶,亦被称为血小板活化因子乙酰水解酶(paf-ah)。lp-pla2随低密度脂蛋白(ldl) 移动,并迅速裂解从ldl氧化得到的氧化的磷脂酰胆碱分子。lp-pla2水解氧化的磷脂酰胆碱的sn-2酯得到脂质介体溶血磷脂酰胆碱(lysopc)和氧化的非酯化脂肪酸(nefa)。文献报道lysopc和nefa能引起炎性反应,因此lp-pla2介导了体内的氧化炎性反应。(zalewski a 等人,arterioscler. thromb. vasc.biol., 25, 5, 923-31 (2005))。
3.文献(wo96/13484、wo96/19451、wo97/02242、wo97/12963、wo97/21675、wo97/21676、wo97/41098、wo97/41099、wo99/2442、wo00/10980、wo00/66566、wo00/66567、wo00/68208、wo01/60805、wo02/30904、wo02/30911、wo03/015786、wo03/016287、wo03/041712、wo03/042179、wo03/042206、wo03/042218、wo03/086400、wo03/87088、wo08/04886、us2008/0103156、us2008/0090851、us2008/0090852、wo08/048866、w005/003118、w006/063811、w006/063813、wo2008/141176、wo2013013503a1、wo2013014185a1、wo2014114248a1、wo2014114694a1、wo2016011930a1、jp200188847 us2008/0279846a1、us 2010/0239565a1、us 2008/0280829a1)描述了许多的lp-pla2抑制剂和/或其用途,治疗涉及血管内皮功能异常或与其相关的疾病、涉及与lp-pla2活性关联的(例如,与溶血磷脂酰胆碱和氧化的游离脂肪酸的形成相关的)脂质氧化的疾病及涉及活化的单核细胞、巨噬细胞或淋巴细胞的或者与单核细胞、巨噬细胞或淋巴细胞的参与增加相关的疾病。具体疾病的例子包括神经退行性疾病(例如阿兹海默症、帕金森病、亨廷顿病、血管性痴呆)、各种神经精神疾病如精神分裂症和自闭症、外周以及脑动脉粥样硬化、中风、代谢性骨病(例如骨髓异常)、血脂异常、佩吉特病、ii型糖尿病、高血压、心绞痛、心肌梗塞、缺血、再灌注损伤、代谢综合征、胰岛素抵抗和甲状旁腺功能亢进、糖尿病性并发症(例如黄斑水肿、糖尿病性视网膜病变和后葡萄膜炎、糖尿病溃疡和糖尿病肾病)、糖尿病外周神经病变性疼痛、炎性疼痛、神经病理性痛、各类癌症(例如前列腺癌、结肠癌、乳腺癌、肾癌、肺癌和卵巢癌等)、黄斑水肿、伤口愈合、男性勃起障碍、类风湿性关节炎、慢性阻塞性肺病(copd) 、败血症、急慢性炎症、牛皮癣和多发性硬化。
4.科学研究结果进一步证明lp-pla2抑制剂可以用于治疗动脉粥样硬化。wilensky等人在加速冠状动脉粥样硬化的糖尿病和高胆固醇血症猪模型中证明lp-pla2抑制剂对于动脉粥样硬化斑块成分的作用(wilensky等人,nature medicine,10,1015-1016 (2008))。临床研究也发现lp-pla2抑制剂能稳定动脉粥样硬化斑块的病人的硬化斑块,阻止斑块进一步发展而发生破裂(serruys等人,circulation 118:1172-1182 (2008))。
5.研究表明高lp-pla2活性与高痴呆症(包括阿尔茨海默病(ad)和混合性痴呆)风险
相关(vanoijen等人,annalsofneurology,59,139(2006);fitzpatrick等人,atherosclerosis235:384-391(2014))。在ad患者中观察到较高的氧化ldl的水平(kassner等人,currentalzheimerresearch,5,358-366(2008);dildar等人,alzheimerdisassocdisord,24,april-june(2010);sinem等人,currentalzheimerresearch,7,463-469(2010))。
6.此外,us2008/0279846描述了lp-pla2抑制剂减少了血脑屏障渗漏和大脑淀粉样蛋白(abeta)负载,可以用于治疗与血脑屏障渗漏相关的疾病,例如阿尔茨海默病和血管性痴呆。在临床研究中,lp-pla2抑制剂对阿尔茨海默病人能起到阻止认知功能进一步恶化的显著效果(maher-edwards等人,alzheimer’s&dementia:translationalresearch&clinicalinterventions1,131-140(2015))。
7.神经炎症(包括多种细胞毒性细胞因子的释放)为所有神经退行性疾病(包括多发性硬化、肌萎缩性侧索硬化、帕金森病、阿尔茨海默病等)的共同特征(perry,actaneuropathol,120,277-286(2010))。lp-pla2抑制剂通过抑制lysopc产生来减少多种细胞因子的释放(shi等人,atherosclerosis191,54-62(2007))。因此,抑制lp-pla2是神经退行性疾病(包括多发性硬化、肌萎缩性侧索硬化、帕金森病等)的潜在治疗方法。
8.lysopc也参与白细胞活化、诱导细胞凋亡和介导血管内皮细胞功能障碍(wilensky等人,currentopinioninlipidology,20,415-420,(2009))。因此,据认为lp-pla2抑制剂可以用于通过减少lysopc的产生而治疗与糖尿病相关的组织损伤。高lp-pla2活性与高糖尿病视网膜病变发病风险有相关性(siddiqui等人,diabetologia,61,1344-1353(2018))。lp-pla2抑制剂可以抑制糖尿病大鼠模型的视网膜病变的主要病理变化(canning等人,p.n.a.s.113,7213-7218(2016)。临床研究也显示lp-pla2抑制剂可以改善糖尿病视网膜病变病人的视网膜黄斑水肿症状和视力(staurenghi等人,ophthalmology122,990-996(2015)。这些研究证明lp-pla2抑制剂可以用于糖尿病视网膜病变。
9.研究表明糖尿病病人体内的lp-pla2活性高于正常人(serban等人j.cell.mol.med.6:643-647,(2002);garg等人indianj.med.res.141:107-114,(2015))。而如上所述,lp-pla2的活性介导了氧化炎性反应,推测抑制lp-pla2活性可以用于治疗糖尿病病人由于体内氧化炎性反应引起的各种并发症,如糖尿病肾病、糖尿病周围神经病变和糖尿病皮肤溃烂等。
10.青光眼和年龄相关性黄斑变性(amd)为视网膜神经退行性疾病。炎症在青光眼和amd的发病机制中起重要作用(buschini等人,progressinneurobiology,95,14-25(2011);tezel,progressinbrainresearch,vol.173,issn0079—6123,第28章)。因此,lp-pla2抑制剂可以提供对于青光眼和amd的潜在治疗应用。
11.在男性勃起障碍病人中,体内lp-pla2的活性显著高于正常人,并认为高lp-pla2活性可以预测早期男性勃起障碍(otunctemur等人,andrologia47:706-710(2015)),提示抑制剂可用于治疗男性勃起障碍。
12.在前列腺癌组织中有lp-pla2高表达,降低lp-pla2可以在体外实验中减少前列腺细胞癌变并促进前列腺癌细胞凋亡(vainio等人,oncotarget,2:1176-1190(2011)),提示lp-pla2抑制剂可以用于治疗前列腺癌。
13.因此,lp-pla2抑制剂可以治疗或预防与lp-pla2活性抑制相关的各种疾病。本发明发现了一种新的lp-pla2抑制剂。
技术实现要素:
14.本发明的一个目的在于提供通式(i)所示的化合物、其顺反异构体、对映异构体、非对映异构体、外消旋体、溶剂合物、水合物、或其药学上可以接受的盐或酯、或其前体药物。
15.本发明的另一个目的在于提供通式(i)所示化合物的制备方法。
16.本发明的另一个目的在于提供通式(i)所示化合物作为lp-pla2抑制剂的用途,从而在制备用于预防、治疗或改善与lp-pla2抑制有关的疾病的药物中的应用,所述疾病如糖尿病并发症、神经炎症相关疾病或/和动脉粥样硬化,所述糖尿病并发症为糖尿病视网膜病变/糖尿病黄斑水肿、糖尿病肾病、糖尿病神经病变、糖尿病外周神经病变性疼痛或/和糖尿病足,神经炎症相关疾病为阿兹海默症、多发性硬化、肌萎缩性侧索硬化或/和帕金森病。
17.本发明的另一个目的在于提供一种药物组合物,其包含一种或多种有效治疗剂量的通式(i)所示化合物、其顺反异构体、对映异构体、非对映异构体、外消旋体、溶剂合物、水合物、或其药学上可以接受的盐或酯、或其前体药物,以及药学上可以接受的载体和/或辅助剂。
18.本发明的另一个目的在于提供一种预防、治疗或改善与lp-pla2抑制有关的疾病的方法,包括给予本发明所述的通式(i)所示化合物、其顺反异构体、对映异构体、非对映异构体、外消旋体、溶剂合物、水合物、或其药学上可以接受的盐或酯、或其前体药物或本发明所述的组合物。
19.在第一方面,一种式i所示的化合物、其顺反异构体、其对映异构体、其非对映异构体、其外消旋体、其溶剂合物、其水合物或其药学上可以接受的盐或其前体药物,其中m为1或2;优选地,m为1;n,u为0,1或2;优选地,n,u为0或1;q为-o-,-s-或-nr
a-,优选的q为-o-;ra为h,c
1-6
烷基,c
1-3
卤代烷基,c
3-8
环烷基或3-8元杂环基;r1为h,卤素,氰基,氨基,c
1-6
烷基,c
1-6
烷氧基,c
1-6
烷基氨基,c
3-8
环烷基或3-8元杂环基,r1可以被一个或多个以下取代基取代:卤素,氰基,c
1-6
烷氧基,c
3-8
环烷基、3-8元杂环基或3-8元杂芳基;(r2)
p 代表环上的氢被p个r2取代,每个r2相同或者不同;
p为2,3,4,5或6;r2,r
x
,ry独立地选自以下取代基:h,卤素,羟基,羧基,氰基,c
1-6
烷基,c
1-6
烷氧基,c
3-8
环烷基,3-8元杂环基,c
6-10
芳基,3-8元杂芳基,-c(o)nrbrc,-s(o)2nrbrc,并且可以被一个或多个以下取代基取代:卤素,氰基,c
1-6
烷氧基,c
3-8
环烷基、3-8元杂环基或3-8元杂芳基;r
x
,ry连同它们连接的碳原子能够形成3-6元饱和环,该环为全碳环或包含一个或多个选自n,o和s原子的杂环,并且可以被一个或多个rm取代;在被p个独立地r2取代的环上,至少有两个r2连接在同一碳原子上,且连同它们共同连接的碳原子形成3-6元饱和环,该环为全碳环或包含一个或多个选自n,o和s原子的杂环,并且可以被一个或多个rm取代;rm为c
1-6
烷基、c
1-3
卤代烷基、卤素,氰基,-orc,-nrbrc,c
3-6
环烷基、3-8元杂环基、c
6-10
芳基或3-8元杂芳基;rb为h,c
1-6
烷基,c
3-8
环烷基或3-8元杂环基;rc为l,l-c(o)-,l-ch
2-或l-s(o)
2-,其中l为h,c
1-6
烷基,c
3-6
环烷基,3-8元杂环基,c
6-10
芳基或3-8元杂芳基,l可以被一个或多个以下基团取代:卤素,羟基,c
1-6
烷氧基,氰基,c
3-8
环烷基,3-8元杂环基,c
6-10
芳基或3-8元杂芳基;a为z为n或cr3;优选地,z为cr3;z’为n或cr4;优选地z’为cr4;r3, r4, r5, r6独立地为h,cn,卤素、c
1-3
烷基或c
1-3
卤代烷基;v为n或cr9,优选地,v为cr9,其中r9为h,cn,卤素,c
1-3
烷基,c
1-3
卤代烷基或-o-w;w为5 或6元杂芳基或苯基,优选为吡啶基、嘧啶基、吡唑基或苯基,其可以任选由一个或多个下列的取代基取代:卤素,氰基,c
1-6
烷基,c
1-3
烷氧基,c
1-3
卤代烷基和c
1-3
卤代烷氧基。
具体实施方式
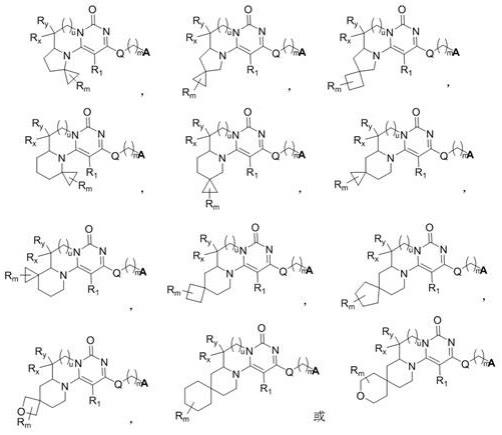
20.在优选的实施方式中,所述式(i)化合物具有以下12个结构中的一个:
其中m为1或2;优选地,m为1;q为-o-,-s-或-nr
a-;优选的q为-o-;ra为h,c
1-6
烷基,c
1-3
卤代烷基,c
3-8
环烷基或3-8元杂环基;r
x
,ry独立地选自以下取代基:h,卤素,羟基,羧基,氰基,c
1-6
烷基,c
1-6
烷氧基,c
3-8
环烷基,3-8元杂环基,c
6-10
芳基,3-8元杂芳基,-c(o)nrbrc,-s(o)2nrbrc,并且可以被一个或多个以下取代基取代:卤素,氰基,c
1-6
烷氧基,c
3-8
环烷基、3-8元杂环基或3-8元杂芳基;r
x
,ry连同它们连接的碳原子能够形成3-6元饱和环,该环为全碳环或包含一个或多个选自n,o和s原子的杂环,并且可以被一个或多个以下取代基取代:c
1-6
烷基、c
1-3
卤代烷基、卤素,氰基,氧代(=o),-orc,-nrbrc,c
3-6
环烷基、3-8元杂环基、c
6-10
芳基或3-8元杂芳基;u为0或1,优选的u为0,同时r
x
,ry均为h;r1为h,卤素,氰基,氨基,c
1-6
烷基,c
1-6
烷氧基,c
1-6
烷基氨基,c
3-8
环烷基或3-8元杂环基,r1任选被一个或多个以下取代基取代:卤素,氰基,c
1-6
烷氧基,c
3-8
环烷基、3-8元杂环基或3-8元杂芳基;优选地,r1为h,卤素,氰基,c
1-6
烷基或c
1-6
烷氧基;优选地,r1为h,卤素,氰基,c
1-3
烷基或c
1-3
烷氧基;更优选地,r1为h,氟,氯,氰基,甲基,乙基或甲氧基;最优选地,r1为h或甲氧基;rm为c
1-6
烷基、c
1-3
卤代烷基、卤素,氰基,氧代(=o),-orc,-nrbrc,c
3-6
环烷基、3-8元杂环基、c
6-10
芳基或3-8元杂芳基;rb为h,c
1-6
烷基,c
3-8
环烷基或3-8元杂环基;
rc为l,l-c(o)-,l-ch
2-或l-s(o)
2-,其中l为h,c
1-6
烷基,c
3-6
环烷基,3-8元杂环基,c
6-10
芳基或3-8元杂芳基,l任选被一个或多个以下基团取代:卤素,羟基,c
1-6
烷氧基,氰基,c
3-8
环烷基,3-8元杂环基,c
6-10
芳基或3-8元杂芳基;a为z为n或cr
3;
优选地,z为cr3;z’为n或cr4;优选地z’为cr4;r3, r4, r5, r6独立地为h,cn,卤素或c
1-3
卤代烷基;v为n或cr9,优选地,v为cr9,其中r9为h,cn,卤素,c
1-3
烷基,c
1-3
卤代烷基或-o-w;w为5 或6元杂芳基或苯基,优选为吡啶基、嘧啶基、吡唑基或苯基,其可以任选由一个或多个以下的取代基取代:卤素,氰基,c
1-6
烷基,c
1-3
烷氧基,c
1-3
卤代烷基和c
1-3
卤代烷氧基;在式(i)和上述12个结构式中,优选地,a为r5, r
6, r7, r8, r9独立地为h、f或cn;或a为r5, r
6, r7, r8独立地为h、f或cn;r9为-o-w;w为5 或6元杂芳基或苯基,优选为吡啶基、嘧啶基、吡唑基或苯基,其可以任选由一个或多个下列的取代基取代: c
1-3
卤代烷基、c1‑ꢀ3卤代烷氧基、cn、卤素和c
1-6
烷基。
21.更优选地,a为r7, r8独立地为h、f或cn;
r9为-o-w;w为吡啶基、嘧啶基、吡唑基或苯基,其可以任选由一个或多个独立选自以下的取代基取代:卤素、cn、cf3、-ocf3、chf2和ch3;最优选地,a选自下列基团:。
22.在一个实施方案中,所述的化合物为以下化合物任一个化合物:
23.上述式的化合物、其盐(例如,药学上可接受的盐)可以立体异构体形式存在(例如,它包含一个或多个不对称碳原子)。所述单个立体异构体(对映异构体和非对映异构体)及它们的混合物均包括在本发明的范围内。
24.本发明还包括上述式的化合物、其盐(例如,药学上可接受的盐)的各种氘代形式。连接到碳原子上的各可用的氢原子可以独立地被氘原子取代。本领域的普通技术人员将了解如何合成上述式的化合物、其盐(例如,药学上可接受的盐)的氘代形式。市售的氘代原料可以用于氘代形式的上述式的化合物、其盐(例如,药学上可接受的盐)的制备中,或可以使用采用氘代试剂(如氘化铝锂)的常规技术来合成这些化合物。
25.除了本技术所述化合物的游离碱或游离酸形式,化合物的盐形式也在本发明的范围内。本发明化合物的盐或药学上可接受的盐可以在化合物的最后分离和纯化过程中在原位制备,或通过单独将游离酸或游离碱形式的纯化的化合物分别与合适的碱或酸反应来制备。对于合适的药学上的盐的综述参见berge等人,j. pharm, sci.,66,1-19,1977; p l gould, international journal of pharmaceutics,33 (1986) ,201-217; 和bighley等人,encyclopedia of pharmaceutical technology, marcel dekker inc,new york 1996, volume 13,page 453-497。
26.本技术所描述的化合物、其盐(例如,药学上可接受的盐)、氘化形式、其溶剂化物或水合物可以以一种或多种多晶型物形式存在。因此,在另一个方面,本发明提供了本技术所定义的化合物、其盐(例如,药学上可接受的盐)的多晶型物,或本技术所述的化合物或其盐(例如,药学上可接受的盐)的溶剂化物或水合物的多晶型物。
27.本发明还包括同位素标记的化合物和盐,其等同于上述式的化合物或其盐,但一个或多个原子被原子质量或质量数不同于自然中最经常发现的原子质量或质量数的原子代替。可掺入上述式的化合物或其盐的同位素的实例为氢、碳、氮、氘的同位素,如3h、
11
c、
14
c和
18
f这些同位素标记的上述式的化合物或其盐可用于药物和/或底物组织分布测试。例如,
11
c和
18
f 同位素可用于pet (正电子发射断层摄影术)。pet可用于脑成像。在一个实施方案中,上述式的化合物或其盐为非同位素标记的。
28.因此,本发明的化合物包括上述式的化合物,或其盐,例如其药学上可接受的盐。本发明代表性化合物包括所述具体的化合物。
29.本发明还涉及包含本发明的化合物和药学上可接受的赋形剂的药物组合物。
30.本发明还涉及治疗或预防与lp-pla2的活性相关的疾病的方法,包括向有需要的受试者给药治疗有效量的本技术所述的本发明的化合物。该疾病可以与以下相关:单核细胞、巨噬细胞或淋巴细胞的参与增加溶血磷脂酰胆碱和氧化的游离脂肪酸的形成与lp-pla2活性关联的脂质的氧化或内皮功能障碍。
31.本发明还提供通过抑制lp-pla2活性治疗或预防疾病的方法。示例性的疾病包括但不限于:神经退行性疾病(例如阿兹海默症、帕金森病、亨廷顿病、血管性痴呆)、各种神经精神疾病如精神分裂症和自闭症、外周以及脑动脉粥样硬化、中风、代谢性骨病(例如骨髓异常)、血脂异常、佩吉特病、ii型糖尿病、高血压、心绞痛、心肌梗塞、缺血、再灌注损伤、代谢综合征、胰岛素抵抗和甲状旁腺功能亢进、糖尿病性并发症(例如黄斑水肿、糖尿病性视网膜病变和后葡萄膜炎、糖尿病溃疡和糖尿病肾病)、糖尿病外周神经病变性疼痛、炎性疼痛、神经病理性痛、各类癌症(例如前列腺癌、结肠癌、乳腺癌、肾癌、肺癌和卵巢癌等)、黄斑水肿、伤口愈合、男性勃起障碍、类风湿性关节炎、慢性阻塞性肺病(copd) 、败血症、急慢性炎症、牛皮癣和多发性硬化。该方法包括向有需要的受试者给药治疗有效量的本发明的化合物。本发明并不意图限制于疾病的任何特定阶段(如早期或晚期)。
32.本发明还提供治疗或预防阿兹海默症的方法。该方法包括向有需要的受试者给药治疗有效量的本发明的化合物。
33.本发明还提供治疗或预防动脉粥样硬化的方法。该方法包括向有需要的受试者给药治疗有效量的本发明的化合物。
34.本发明还提供了通过给药本发明的化合物治疗或预防眼部疾病的方法。在一些实施方式中,本发明提供了治疗黄斑水肿的方法,其包括向受试者给药治疗有效量的本发明的化合物。在一些实施方式中,该黄斑水肿与糖尿病性眼病(例如糖尿病性黄斑水肿或糖尿病性视网膜病)有关。在一个实施方式中,该黄斑水肿与后葡萄膜炎有关。
35.本发明还提供了本发明的化合物在制备用于治疗或预防本技术所述的疾病的药物中的用途。
36.本发明还提供了本发明化合物,其用于本技术所述的治疗或预防。
37.本技术使用的“和/或”是指包括一个或多个关联的列举项目的任何和所有可能的组合。可以进一步理解,本说明书中使用的术语“包含”和/或“包括”指明所指的特征、整体、步骤、操作、元素和/或组分的存在,但不排除一个或多个其他特征、整体、步骤、操作、元素、组分和/或其组合的存在或加入。
38.一般而言,本技术使用的命名和本技术所描述的有机化学、药物化学、生物学的实验操作是本领域公知的,而且在本领域中普遍采用。除非另有定义,本技术使用的所有技术和科学术语一般具有本公开所属领域的普通技术人员通常理解的相同含义。在本技术使用的术语存在多个定义的情况下,除非另有说明,以本部分的定义为准。
39.定义如本技术使用的,除非另有所述,术语“疾病”是指机体或一些器官的状态的任何改变,其中断或干扰功能的执行和/或引起患病的人或与其接触的人的症状(如不适、功能障碍、不良应激乃至死亡)。
40.如本技术使用的,除非另有所述,“糖尿病视网膜病变”是指糖尿病导致的慢性进展性视网膜微血管渗漏和梗阻的结果。“糖尿病性黄斑水肿”是指由于糖尿病引起的黄斑中心凹一个视盘直径范围内的细胞外液积聚所致的视网膜增厚或硬性渗出沉积。
41.如本技术使用的,除非另有所述,“神经退行性疾病”是指特征为神经组织和/或神经组织功能的逐步的、进行性损失的不同种类的中枢神经系统障碍。神经退行性疾病为一类神经系统疾病,其中神经系统疾病的特征为神经组织的逐步的、进行性损失和/或改变的神经功能,通常为由于逐步的、进行性的神经组织损失而导致神经功能的降低。在一些实施方式中,本技术所述神经退行性疾病包括其中存在有缺陷的血脑屏障(例如渗透性血脑屏障)的神经退行性疾病。其中存在缺陷性血脑屏障的神经退行性疾病的例子包括但不限于,阿兹海默症、亨廷顿病、帕金森病和血管性痴呆等。
42.如本技术使用的,除非另有所述,“血管性痴呆”也被称为“多发性梗塞性痴呆",其是指一组由不同的机制(其都导致脑中的血管损害)引起的综合征。例如,血管性痴呆的主要亚型为血管性轻度认知功能障碍、多发性梗塞性痴呆、由于重大的单梗塞(影响丘脑、大脑前动脉、顶叶或扣带回)导致的血管性痴呆、由于出血性损害导致的血管性痴呆、小血管疾病(包括,例如由于腔隙性损害和宾斯旺格病(binswanger disease) 导致的血管性痴呆)及混合型痴呆。
43.如本技术使用的,除非另有所述,“神经病理性痛”是为由神经系统原发性损害和功能障碍所激发或引起的疼痛。
44.如本技术使用的,除非另有所述,“炎性疼痛”是局部急性炎症或是慢性炎症刺激神经所致的疼痛。
45.如本技术使用的,除非另有所述,“糖尿病外周神经病变性疼痛”是指糖尿病并发的神经损伤而引起的疼痛,糖尿病中的神经损伤至少部分是由于血流减少和高血糖引起的。
46.如本技术使用的,除非另有所述,“血脑屏障”或"bbb" 在本技术可互换使用,用于指存在于穿过脑组织的血管中的可渗透屏障,其严格地限制和密切地调节物质在血液和脑组织之间的交换。血脑屏障组分包括形成所有血管的最内层衬里的内皮细胞、作为bbb的结构性关联物的相邻内皮细胞之间的紧密连接物、内皮细胞的基底膜和覆盖几乎所有暴露的血管外表面的附近星形胶质细胞的扩大的足突。
47.如本技术使用的,除非另有所述,“代谢性骨病”是指特征为骨组织的逐步和进行性损失的不同种类的骨疾病。本技术所述的代谢性骨病为其中存在扩散性骨密度下降和/或骨强度降低的状况的骨代谢疾病。这类疾病通过组织学外观来表征。示例性的代谢性骨病包括但不限于,特征为矿物质和骨基质减少的骨质疏松症和特征为矿物质减少但具有完整的骨基质的骨软化症。
48.如本技术使用的,除非另有所述,“骨质减少疾病”或“骨质减少”可以在本技术中互换使用,涉及具有下降的钙化和/或骨密度的状况,为用来表示在其中观察到钙化和/或骨密度下降的所有骨骼系统的描述性术语。骨质减少也是指由于类骨质(osteoid) 合成不足导致的骨质减少。
49.如本技术使用的,除非另有所述,“骨质疏松症”是指其中矿物质和/或骨基质减少和/或骨基质减少的病症。
50.如本技术所用,除非另有所述,“烷基”是具有指定碳原子数的一价、饱和经链。例如,c
1-3
烷基是指具有1 至3个碳原子的烷基。c
1-6
烷基是指具有1至6个碳原子的烷基。烷基可为直链或支链的。在一些实施方案中,支链的烷基可具有一个、两个或三个支链。示例性烷基包括,但不限于,甲基、甲基乙基、乙基、丙基(正丙基和异丙基)、丁基(正丁基、异丁基和叔丁基)。
51.如本技术所用,除非另有所述,“烷氧基”取代基为式“r-o
‑”
的基团,其中r为如上定义的烷基。例如, c
1-3
烷氧基是指包含1 至3个碳的这种烷氧基取代基。示例性烷氧基取代基包括但不限于,甲氧基、乙氧基、正丙氧基、正丁氧基、正戊氧基、正已基氧基、异丙氧基、异丁氧基、仲丁氧基、叔丁氧基、异戊氧基和新戊氧基。
52.如本技术所用,除非另有所述,“环烷基”是指单价饱和的环状烃基包括桥环及螺环,优选具有3-7个环碳原子,例如环丙基、环丁基、环戊基或[1.1.1]螺桨烷,以及下文中特别示例的那些基团。
[0053]
如本技术所用,除非另有所述,“芳基”表示包含一个或多个芳环的烃基,如苯基或萘基等。
[0054]
如本技术所用,除非另有所述,“杂芳基”表示在各环中具有至多8个原子的稳定单环、双环、或三环,其中至少一个环是芳族的并且至少一个环含有1至4个选自o、n 和s 的杂
原子。在该定义的范围内的杂芳基包括但不限于吖啶基、咔唑基、噌啉基、喹喔啉基、喹唑啉基、吡唑基、吲哚基、异吲哚基、1h, 3h-1-氧代异吲哚基、苯并三唑基、呋喃基、噻吩基、吡啶并吗啉基、吡啶并哌啶基、吡啶并吡咯烷基、苯并噻吩基、苯并呋喃基、苯并二噁烷、苯并二氧杂苯、喹啉基、异喹啉基、噁唑基、异噁唑基、苯并噁唑基、咪唑基、吡嗪基、哒嗪基、吡啶基、嘧啶基、吡咯基、四氢喹啉基、噻唑基、异噻唑基、1, 2, 3-三唑基、1, 2, 4—三唑基、1, 2, 4-噁二唑基、1, 2, 4-噻二唑基、1, 3, 5-三嗪基、1, 2, 4-三嗪基、1, 2, 4, 5-四嗪基、四唑基、呫吨基、吩嗪基、吩噻嗪基、吩噁嗪基、氮杂
䓬
基、氧杂
䓬
基和硫杂
䓬
基。特别的杂芳基具有5 或6 元环,例如呋喃基、吡啶基、哒嗪基、嘧啶基、吡嗪基、噻吩基、异唑基、噁唑基、二唑基、咪唑基、吡咯基、吡唑基、三唑基、四唑基、噻唑基、异噻唑基、噻二唑基,吡啶并吗啉基、吡啶并哌啶基、吡啶并吡咯烷基。
[0055]
如本技术所用,除非另有所述,“杂环”或“杂环基”是指1至4个碳原子已被独立地选自n、n(r)、s、s(o)、s(o)和o的杂原子代替的环烃。杂环可以是饱和或不饱和的,但不是芳族的。杂环基也可以是含有1、2 或3 个环,包括桥环及螺环结构。适合的杂环基的实例包括但不限于:氮杂环丁烷、氧杂环丁烷、四氢呋喃基、四氢噻吩基、吡咯烷基、2-氧代吡咯烷基、吡咯啉基、吡喃基、二氧戊环基、哌啶基、2-氧代哌啶基、吡唑啉基、咪唑啉基、噻唑啉基、二硫杂环戊二烯基、氧杂硫杂环戊二烯基、二噁烷基、二噁烯基、二噁唑基、噁噻唑基(oxathiozolyl) 、噁唑酮基、哌嗪基、吗啉代、硫代吗啉基、3-氧代吗啉基、二噻烷基、三噻烷基和噁嗪基。
[0056]“桥环化合物”术语指一个或多个原子(即c、o、n或s) 连接两个不相邻的碳原子或氮原子。优选的桥环包括但不限于:一个碳原子、两个碳原子、一个氮原子、两个氮原子和一个碳-氮基。值得注意的是,一个桥总是将单环转换成三环。桥环中,环上的取代基也可以出现在桥上。
[0057]“螺环化合物”术语是指两个单环共用一个碳原子的多环化合物,共用的碳原子称为螺原子。
[0058]
如本技术所用,除非另有所述,“卤素”是指氟(f)、氯(cl)、溴(br)或碘(i)。卤代是指卤素基团:氟(-f)、氯(-cl)、溴(-br)、或碘(
‑ꢀ
i)。
[0059]
如本技术所用,除非另有所述,“卤代烷基”为取代有一个或多个卤素取代基的烷基,该卤素取代基可相同或不同。例如,c
1-3
卤代烷基是指包含1 至3个碳的卤代烷基取代基。示例性卤代烷基取代基包括,但不限于,单氟甲基、二氟甲基、三氟甲基、1-氯-2-氟乙基、三氟丙基、3-氟丙基和2-氟乙基。
[0060]
如本技术所用,除非另有所述,当环上两个取代基连同它们的互相连接的原子一起结合形成另一环时,该环可为螺环稠合或单边稠合的。螺环-稠合的环体系由两个环组成,该两个环仅具有一个共同的碳原子。单边-稠合的环体系由两个环组成,该两个环仅共享两个原子和一个键。
[0061]
如本技术所用,除非另有所述,“任选取代的”表示基团或环可为未取代的,或该基团或环可取代有一个或多个本技术定义的取代基。
[0062]
如本技术所用,除非另有所述,“4、5或6元饱和环,该环任选包含一个选自n或o的杂原子”是指4、5或6元饱和碳环且一个碳原子环成员可任选被一个选自n或o的杂原子代替,例如、环丁基、环戊烷基、环己烷基、氮杂环丁烷基(azitidinyl)、吡咯烷基、哌啶基、氧
杂环丁烷基、四氢呋喃基和四氢-2h-吡喃基。
[0063]
如本技术所用,除非另有所述,涉及疾病的“治疗”、“治”或“处理”是指: (1)减轻疾病或减轻疾病的一种或多种生物学表现,(2)干扰 (a)导致或造成疾病的生物学级联中的一个或多个点或(b)疾病的一种或多种生物学表现,(3)缓和与疾病相关的一种或多种症状或效应,和/或(4)减缓疾病的进展或疾病的一种或多种生物学表现,和/或(5)减少疾病严重性或疾病生物表现的可能性。
[0064]
如本技术所用,除非另有所述,“预防”是指预防性给药药物以减少疾病或其生物表现的发生的可能性或延迟该发生。
[0065]
如本技术所用,除非另有所述,“受试者”是指哺乳动物受试者(例如,狗、猫、马、牛、绵羊、山羊、猴等),和尤其是人类受试者。
[0066]
如本技术所用,除非另有所述,“药学上可接受的盐”是指保留所述主题化合物所需的生物活性,并显示出最低的不希望的毒理效应的盐。这些药学上可接受的盐可以在化合物的最后分离和纯化过程中原位制备,或通过单独地将游离酸或游离碱形式的纯化化合物分别与合适的碱或酸反应来制备。
[0067]
如本技术所用,除非另有所述,术语“治疗有效量”是指相比于未接受该量的相应受试者,导致治疗或预防疾病的量,但该量在合理医学判断的范围内足够低以避免严重副作用(以合理的益处/风险比)。化合物的治疗有效量将随着选择的具体化合物(例如考虑到化合物的效力、功效和半衰期):选择的给药途径;治疗的疾病;治疗的疾病的严重性;治疗患者的年龄、体型、体重和身体状况:治疗患者的医疗史;治疗的持续时间;并行治疗的性质;所需治疗效果等因素而改变,但仍可通过本领域技术人员以常规方式确定。
[0068]
化合物合成本领域的技术人员可以理解,如果本技术描述的取代基与本技术描述的合成方法不相容,该取代基可以用在反应条件下稳定的合适的保护基进行保护。保护基可以在反应顺序中合适的点脱除,以得到所需的中间体或目标化合物。合适的保护基和使用这类合适保护基对不同的取代基进行保护和去保护的方法对于本领域技术人员是公知的;这方面的例子可以在i.greene 和p.wuts, protecting groups in chemical synthesis (第三版), johnwiley&sons , ny (1999) 中找到。在一些情况下,可以具体地选择在使用的反应条件下具有反应性的取代基。在这些情况下,反应条件转化所选择的取代基成为可用作中间化合物的另一种取代基或者为目标化合物中的需要的取代基的另一种取代基。
[0069]
一般方案一般方案提供式1.8化合物的一般合成路线,其中r1,r2,r
x
,ry,q, m, n, p, u, a如关于式(i)中所定义。所述路线包括以下步骤:步骤(i):通过将化合物(1.1)脱除boc保护基生成化合物(1.2);步骤(ii): 将化合物(1.2)与化合物(1.3)反应生成化合物(1.5);步骤(iv):通过将化合物(1.5)关环得到化合物(1.7);步骤(v): 化合物(1.7)与hq-(ch2)
m-a反应得到最终产物(1.8),即为式i的化合物;或包含以下步骤:步骤(i):通过将化合物(1.1)脱除boc保护基生成化合物(1.2);
步骤(iii):将化合物(1.2)与化合物(1.4)反应生成化合物(1.6);步骤(iv’):通过将化合物(1.6)关环得到化合物(1.7);步骤(v): 化合物(1.7)与hq-(ch2)
m-a反应得到最终产物(1.8),即为式i的化合物;其中,r1,r2,r
x
,ry,q, m, n, p, u, a如本技术中所定义。
[0070]
具体地,最终化合物1.8的制备可以通过i, ii, iv, v的步骤也可以通过i, iii, iv, v的步骤获得。步骤(i)可以通过将化合物1.1在合适的酸性试剂,例如氯化氢/1,4-二氧六环溶液中,在合适温度,例如室温,脱除boc保护基生成化合物1.2。步骤(ii)或(iii)可作为sn
ar
反应,使用合适的碱性试剂,例如三乙胺,在合适的溶剂,例如乙腈中,在合适的温度,例如室温,将化合物1.2与1.3反应生成化合物1.5或者将化合物1.2与1.4反应生成化合物1.6。化合物1.5或1.6均可以通过相同的操作步骤(iv)得到同样的中间体1.7,步骤(iv)可以通过将化合物1.5或1.6与合适试剂,如三乙胺/甲磺酰氯,或者二氯亚砜,在合适的温度如0oc或室温,将羟基转化为甲磺酸酯或氯代,然后不经纯化进一步在碱性试剂,例如碳酸钾或碳酸钠和合适溶剂,例如乙腈中,加热反应,关环得到化合物1.7。步骤(v)将1.7与对应的醇,硫醇,胺hq-(ch2)
m-a (q为-o-,-s-,-nr
a-)在合适的碱如nah条件下在合适的溶剂如乙腈中反应得到最终产物1.8。
[0071]
用途本发明化合物是lp-pla2抑制剂。因此,这些化合物可用于治疗,例如治疗或预防与lp-pla2的活性有关的疾病,其包括使用治疗有效量的lp-pla2抑制剂治疗需要该治疗的受试者。因此,本发明的一方面涉及治疗或预防与lp-pla2活性有关的疾病的方法。本领域技术人员可以理解,特定的疾病或其治疗可以涉及与lp-pla2活性有关的一种或多种潜在机制,包括一种或多种本技术中描述的机制。
[0072]
在一些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防以下公开的专利申请公开任意疾病的药物中的用途:wo96/13484,wo96/19451,wo97/02242,wo97/
12963,wo97/21675,wo97/21676,wo97/41098,wo97/41099,wo99/24420,wo00/10980,wo00/66566,wo00/66567,wo00/68208,wo01/60805,wo02/30904,wo02/30911,wo03/015786,wo03/016287,wo03/041712,wo03/042179,wo03/042206,wo03/042218,wo03/086400,wo03/87088,wo08/048867,us2008/0103156,us2008/0090851,us2008/0090852、wo08/048866、wo05/003118 (ca 2530816al)、wo06/063811 、wo06/063813、wo2008/141176、wo2013013503a1、wo2013014185a1、wo2014114248a1、wo2014114694a1、wo2016011930a1、jp 200188847、us 2008/0279846 a1、us 2010/0239565 a1、和us 2008/0280829 a1。
[0073]
在一些实施方案中,本发明提供本发明的化合物在制备用于治疗眼病的药物中的用途。本发明中适用的眼病可能与血视网膜内屏障(ibrb) 的破坏有关。示例性的眼病涉及糖尿病性眼病,其包括黄斑水肿、糖尿病性视网膜病、后葡萄膜炎、视网膜静脉闭塞等。更多的眼病包括但不限于,视网膜中央静脉阻塞、视网膜分枝静脉阻塞、伊-加综合征(白内障后和手术后)、色素性视网膜炎、平坦部炎、鸟枪弹样视网膜脉络膜病变、视网膜外层膜、脉络膜肿瘤、囊性黄斑水肿、旁中心凹毛细血管扩张、牵引性黄斑病、玻璃体黄斑牵引综合征、视网膜剥离、视神经视网膜炎、特发性黄斑水肿等。使用lp-pla2抑制剂治疗眼病的更详细内容提供在wo2012/080497 中,将其引入本技术作为参考。
[0074]
此外,本发明一些实施方案提供本发明的化合物在制备用于治疗或预防受试者的糖尿病性黄斑水肿的药物中的用途。在一些实施方案中,本发明提供了本发明的化合物在用于治疗受试者的糖尿病性黄斑水肿的用途。
[0075]
在某些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防患有黄斑水肿或具有患黄斑水肿风险的受试者的药物中的用途。在一些实施方案中,本发明提供本发明的化合物在制备用于治疗患有黄斑水肿或具有患黄斑水肿风险的受试者的药物中的用途。在另一个实施方案中,所述黄斑水肿与糖尿病性眼病有关,例如糖尿病黄斑水肿或糖尿病性视网膜病。在另一个实施方案中,所述黄斑水肿与后葡萄膜炎有关。
[0076]
在某些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防青光眼或黄斑变性的药物中的用途。在一些实施方案中,本发明提供本发明的化合物在制备用于治疗青光眼或黄斑变性的药物中的用途。
[0077]
在一个实施方案中,本发明提供本发明的化合物在制备用于治疗或预防需要此治疗的受试者中的与血视网膜内屏障破坏有关的疾病的药物中的用途。在一个实施方案中,本发明提供本发明的化合物在制备用于治疗在需要此治疗的受试者中的与血视网膜内屏障破坏有关的疾病的药物中的用途。
[0078]
在一些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防以下任意疾病的药物中的用途,该疾病涉及内皮机能障碍,例如,动脉粥样硬化(例如,外周血管动脉粥样硬化和脑血管动脉粥样硬化)、糖尿病、高血压、心绞痛、局部缺血和再灌注后的病症。
[0079]
在一些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防以下任意疾病的药物中的用途,该疾病涉及与酶活性相关联的脂质氧化,例如,除了例如动脉粥样硬化和糖尿病等病症外的其他病症,例如类风湿性关节炎、中风、脑的炎性病症(例如阿兹海默症)、各种神经精神病症(如精神分裂症、自闭症)、心肌梗死、局部缺血、再灌注损伤、败血症以及急性和慢性炎症。
[0080]
在一些实施方案中,本发明提供本发明的化合物在制备用于降低患有冠心病的患
者中心血管事件(例如心脏病发作、心肌梗塞或中风)的几率的药物中的用途。
[0081]
在一些实施方案中,本发明提供本发明的化合物在制备用于治疗或预防以下疾病的药物中的用途,该疾病涉及活化的单核细胞、巨噬细胞或淋巴细胞,因为所有这些细胞种类表达lp-pla2,包括涉及活化的巨噬细胞(如m1、枝状和/或其它产生氧化应激的巨噬细胞)的疾病。示例性的病症包括但不限于牛皮癫、类风湿性关节炎、伤口愈合、慢性阻塞性肺病(copd) 、肝硬化、特应性皮炎、肺气肿、慢性胰腺炎、慢性胃炎、主动脉瘤、动脉粥样硬化、多发性硬化、阿兹海默症和自身免疫疾病如狼疮。
[0082]
在其它实施方案中,本发明提供本发明的化合物在制备以下药物中的用途,该药物用于急性冠状动脉事件(例如由动脉粥样硬化引起的)的初级或次级预防;预防再狭窄的辅助治疗;或延迟糖尿病或高血压性肾功能不全的发展。预防包括治疗具有这类病症风险的受试者。
[0083]
在一些实施方案中,本发明提供在需要此治疗的受试者中治疗或预防与异常血脑屏障(bbb) 功能、炎症和/或小神经胶质活化有关的神经系统病症的方法。在一些实施方案中,本发明提供在需要此治疗的受试者中治疗或预防与异常血脑屏障(bbb)功能、炎症和/或小神经胶质活化有关的神经系统病症的方法。所述方法包括将治疗有效量的本发明化合物给药至受试者。在另一个实施方案中,所述异常bbb是渗透性bbb。在另一个实施方案中,所述疾病是神经退行性疾病。这类神经退行性疾病例如是但不限于,血管性痴呆、阿兹海默症、帕金森病和亨廷顿舞蹈病。在一个实施方案中,本发明提供治疗或预防受试者的与血脑屏障(bbb) 泄漏有关的疾病的方法。在一些实施方案中,本发明提供了治疗受试者的与血脑屏障(bbb) 泄漏有关的疾病的方法。示例性的疾病包括但不限于,脑出血、脑淀粉样血管病。在一个实施方案中,所述神经退行性疾病是阿兹海默症。在具体实施方案中,所述神经退行性疾病是血管性痴呆。在一个实施方案中,所述神经退行性疾病是多发性硬化症(ms)。
[0084]
在一个实施方案中,本发明化合物可用于治疗或预防受试者的神经退行性疾病。所述方法包括将本发明化合物(例如为包含本发明化合物的药物组合物的形式)给药于需要此治疗的受试者。在一个实施方案中,本发明化合物可用于治疗受试者的神经退行性疾病。示例性的神经退行性疾病包括但不限于,阿兹海默症、血管性痴呆、帕金森病和亨廷顿舞蹈病。在一具体实施方案中,本发明所述神经退行性疾病与异常的血脑屏障有关。在一个实施方案中,被给药抑制lp-pla2活性的药剂的受试者是人。
[0085]
在一个实施方案中,本发明提供治疗或预防患有血管性痴呆或具有患血管性痴呆风险的受试者的方法。所述方法包括将本发明化合物(例如包含治疗有效量的本发明化合物的药物组合物)给药至受试者。在一个实施方案中,本发明提供治疗患有血管性痴呆或具有患血管性痴呆风险的受试者的方法。在一具体实施方案中,所述血管性痴呆与阿兹海默症有关。
[0086]
在某些实施方案中,本发明涉及通过将治疗有效量的本发明化合物给药于需要此治疗的受试者以治疗或预防代谢性骨病的方法。在一些实施方案中,本发明涉及通过将治疗有效量的本发明化合物给药至需要此治疗的受试者以治疗代谢性骨病的方法。示例性的代谢性骨病包括与骨质和骨密度损失有关的疾病,包括但不限于骨质疏松症和骨质减少疾病。示例性的骨质疏松症和骨质减少疾病包括但不限于,骨髓异常、血脂异常、佩吉特氏病、ii型糖尿病、代谢综合征、胰岛素抵抗、甲状旁腺功能亢进和相关疾病。在另一个实施方案
中,需要此治疗的受试者是人。
[0087]
认为预防本技术描述的骨质疏松症和/或骨质减少疾病的方法可能受到抑制lp-pla2的表达和/或抑制lp-pla2的蛋白活性的影响。因此,本发明的一些实施方案提供通过阻断酶活性来抑制lp-pla2的方法。在另一个实施方案中,提供了通过降低和/或下调lp-pla2rna的表达从而抑制lp-pla2的方法。在另一个实施方案中,预防和/或降低骨质损失和i或骨密度损失导致预防或减少与代谢性骨病例如骨质疏松症和/或骨质减少疾病有关的症状。
[0088]
在具体实施方案中,所述方法还包括将用于治疗代谢性骨病的其他治疗剂给药于需要治疗的受试者。例如,当所述代谢性骨病是骨质疏松症时,可以使用其他治疗剂,例如双磷酸盐类(bisphosphates)(例如,阿仑磷酸盐、伊班磷酸盐、利塞磷酸盐、降血钙素、雷洛昔芬)、选择性雌激素调节剂(serm)、雌激素疗法、激素替代疗法(et/hrt)和特立帕肤。
[0089]
在一个实施方案中,全身性炎性疾病例如幼年型类风湿关节炎、炎性肠病、川崎病、多发性硬化、结节病、多动脉炎、牛皮癣性关节炎、反应性关节炎、系统性红斑狼疮、伏-小柳-原田综合征、莱姆病、贝赫切特病、强直性脊柱炎、慢性肉芽肿性疾病、起止点炎(enthesitis)可能是影响视网膜的后葡萄膜炎的根本原因,且其可导致黄斑水肿。本发明涉及通过给药治疗有效量的本发明化合物治疗或预防后葡萄膜炎或这些全身炎性疾病中的任一种的方法。在一个实施方案中,本发明提供了通过给药治疗有效量的本发明化合物治疗后葡萄膜炎或这些全身炎性疾病中的任一种的方法。
[0090]
与lp-pla2活性相关的疾病的治疗和/或预防可在单一疗法或在双重或多重组合治疗中使用本发明化合物来获得。例如本发明化合物可用于与抗高血脂剂、抗动脉粥样硬化剂、抗糖尿病剂、抗心绞痛剂、抗炎剂或抗高血压剂或用于降低脂蛋白(a)(lp(a))的药剂结合来治疗或预防本发明描述的疾病。上述药剂的例子包括但不限于,胆固醇合成抑制剂,例如他汀类:抗氧化剂,例如丙丁酚;胰岛素致敏剂;钙通道拮抗剂和抗炎药,例如非甾体抗炎药(nsaid)。用于降低lp(a)的药剂包括wo97/02037、wo98/28310、wo98/28311和wo98/28312中所述的氨基磷酸醋。在一个实施方案中,本发明化合物可与一个或多个他汀-起使用。他汀类是众所周知的胆固醇降低剂,包括阿托伐他汀、辛伐他汀、普伐他汀、西立伐他汀、氟伐他汀、洛伐他汀和瑞舒伐他汀。在一些实施方案中,本发明化合物可与抗糖尿病药或胰岛素致敏剂一起使用。在一个实施方案中,本发明化合物可与pparγ激活剂,例如gi262570(glaxosmithkline)和格列酮类(glitazone)化合物例如罗格列酮、曲格列酮和吡格列酮一起使用。该药剂可以例如本领域已知的治疗有效量或以比本领域已知的可提供有效治疗的给药量更小或更多的量给药。
[0091]
组合治疗包括以分开的剂型或在单一剂型中一起给药治疗药剂。组合治疗可包括同时给药或分开给药治疗药剂,其可为基本上同时或基本上分开给药。典型地,组合治疗包括给予每个药剂使得每个药剂的治疗有效量在至少一段重叠的时间内存在于受试者的体内。
[0092]
使用方法本发明化合物的治疗有效量将取决于许多因素,例如包括预期接受者的年龄和体重、需要治疗的精确病症及其严重性、制剂性质和给药途径,且将最终取决于开具处方的医生的判断。然而,用于治疗文中所述疾病的本发明化合物的治疗有效量将通常在0.1到100
pharmaceutical sciences, mack 出版公司)、药物添加剂手册(the hand book of pharmaceutical additives, gower 出版有限公司)和药物赋形剂手册(handbook of pharmaceutical excipients, 美国药物协会和药物出版社)。
[0100]
使用本领域技术人员已知的技术和方法制备本发明药物组合物。常用于本领域的一些方法描述在remington药物科学(mack出版社)中。
[0101]
一个方面,本发明涉及包含治疗有效量的本发明化合物和桸释剂或填充剂的固体口服剂型,例如片剂或胶囊。合适的稀释剂和填充剂包括乳糖、催糖、葡萄糖、甘露醇、山梨醇、淀粉(例如玉米淀粉、马铃薯淀粉和预胶化淀粉)、纤维素及其衍生物(例如微晶纤维素)、硫酸钙和磷酸氢钙。口服固体剂型还可包括粘合剂。合适的粘合剂包括淀粉(例如玉米淀粉、马铃薯淀粉和预胶化淀粉)、明胶、阿拉伯胶、藻酸钠、藻酸、黄胶、瓜尔胶、聚维酮和纤维素及其衍生物(例如微晶纤维素)。口服固体剂型还可以包括含崩解剂。合适的崩解剂包括交聚维酮、淀粉羟基乙酸钠、交联羧甲纤维素(croscarmelose) 、藻酸和羧甲基纤维素钠。口服固体剂型还可以包含润滑剂。适当的润滑剂包括硬脂酸、硬脂酸镁、硬脂酸钙和滑石。
[0102]
在具体实施方式中,本发明涉及含0.01mg至1000mg一种或多种文中所述的上述式的化合物或其药学上可接受的盐以及0.01g至5g的一种或多种药学上可接受的赋形剂的药物组合物。
[0103]
实施例制备中间体1(s)-2-(((叔丁氧羰基)氨基)-6-(二甲基(氧代)-λ
6-亚磺酰基)-5-氧代己酸甲酯室温下,将1-(叔丁基)2-甲基(s)-5-氧吡咯烷-1, 2-二羧酸酯(49 g,0.2 mol),叔丁醇钾(34 g,0.3 mol)加入二甲亚砜(500 ml)中,室温搅拌1小时,加入三甲基碘化亚砜(75 g,0.34 mol),室温搅拌3小时。反应液倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(41 g,61 %)。ms: m/z [m+h]
+ =336。
[0104] 1-(叔丁基)2-甲基(s)-5-氧哌啶-1, 2-二羧酸酯室温下,(s)-2-(((叔丁氧羰基)氨基)-6-(二甲基(氧代)-λ
6-亚磺酰基)-5-氧代己酸甲酯(4.1 g,12 mmol)和ir[cod]2cl(41 mg,0.6 mmol)加入1, 2-二氯乙烷(50 ml)中,氩气保护下,90 o
c搅拌2天。反应液浓缩,柱层析纯化得到标题化合物(1.3 g,33 %)。 ms: m/z [m+h]
+ =258。
[0105] 1-(叔丁基)2-甲基(s)-5-亚甲基哌啶-1, 2-二羧酸酯
室温下,将1-(叔丁基)2-甲基(s)-5-氧哌啶-1, 2-二羧酸酯(500 mg,1.9 mmol)加入无水四氢呋喃(50 ml)中,氩气保护下降温至0 o
c,滴加正丁基锂的正己烷溶液(2.5 m,1 ml,2.5 mmol),0 o
c搅拌1小时。将甲基三苯基溴化磷(500 mg,1.9 mmol)加入反应液,室温搅拌过夜。反应液用水淬灭,二氯甲烷提取,有机相浓缩,柱层析纯化得到化合物(250 mg,50 %)。 ms: m/z [m+h]
+ =256。
[0106] 1-苄基 2-甲基(s)-5-亚甲基哌啶-1, 2-二羧酸酯室温下,将1-(叔丁基)2-甲基(s)-5-亚甲基哌啶-1, 2-二羧酸酯(250 mg,1.0 mmol)和三氟乙酸(228 mg,2.0 mmol)加入二氯甲烷(5 ml)中,室温搅拌1小时。反应液浓缩,向残留物中依次加入水(5 ml)、碳酸钾(414 mg,3.0 mmol)和氯甲酸苄酯(170 mg,1.0 mmol),室温搅拌过夜。反应液倒入水中,二氯甲烷提取,有机相浓缩,制备薄层色谱分离得到标题化合物(110 mg,38 %)。ms: m/z [m+h]
+ =290。
[0107]
5-苄基 6-甲基(s)-5-氮杂螺[2. 5]辛烷-5, 6-二羧酸酯室温下,将二乙基锌的正己烷溶液(1 m,7.6 ml,7.6 mmol)加入二氯甲烷(7 ml)中,氩气保护下降温至0 o
c,将三氟乙酸(560 μl,7.6 mmol)加入反应液,0 o
c搅拌1小时。将二碘甲烷(2.0 g,7.6 mmol)加入反应液,0 o
c搅拌1小时。将1-苄基 2-甲基(s)-5-亚甲基哌啶-1, 2-二羧酸酯(1.1 g,3.8 mmol)加入反应液,室温搅拌过夜。反应液用二氯甲烷稀释后倒入饱和碳酸氢钠水溶液中,室温搅拌0.5小时。过滤,滤液用二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(300 mg,27 %)。ms: m/z [m+h]
+ =304。
[0108] (s)-5-氮杂螺 [2. 5]辛烷-6-羧酸甲酯室温下,将5-苄基 6-甲基(s)-5-氮杂螺[2. 5]辛烷-5, 6-二羧酸酯(300 mg,1.0 mmol)和钯碳(10 %,30 mg)加入甲醇(10 ml)中,室温常压氢化过夜。反应液过滤,滤液浓缩得到标题化合物粗品直接用于下一步反应。ms: m/z [m+h]
+ =170。
[0109] (s)-(5-氮杂螺[2. 5]辛烷-6-基)甲醇
室温下,将(s)-5-氮杂螺[2. 5]辛烷-6-羧酸甲酯(190 mg,1.1 mmol)加入无水四氢呋喃(2 ml)中,搅拌下加入氢化铝锂(45 mg,1.1 mmol),室温搅拌2小时。反应液用十水硫酸钠淬灭,室温搅拌30分钟,过滤,滤饼用四氢呋喃淋洗,滤液浓缩得到标题化合物(100 mg,2步收率:64 %)。ms: m/z [m+h]
+ =142。
[0110] (s)-(5-(2, 6-二氯嘧啶-4-基)-5-氮杂螺[2. 5]辛烷-6-基)甲醇室温下,将(s)-(5-氮杂螺[2.5]辛烷-6-基)甲醇(100 mg,0.7 mmol)和碳酸钠固体(150 mg,1.4 mmol)加入乙腈(2 ml)中,氩气保护下冷却至0 o
c,加入2, 4, 6-三氯嘧啶(130 mg,0.7 mmol),室温搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(100 mg,50 %)。ms: m/z [m+h]
+ =288。
[0111] (s)-3'-氯-8', 9', 9a', 10'-四氢-1'h, 6'h-螺[环丙烷-1, 7'-吡啶[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮室温下,将(s)-(5-(2, 6-二氯嘧啶-4-基)-5-氮杂螺[2. 5]辛烷-6-基)甲醇(50 mg,0.2 mmol)和二氯亚砜(250 mg,0.2 mmol)加入二氯甲烷(2 ml)中,室温搅拌过夜。反应液浓缩,向残留物中依次加入碳酸钾固体(50 mg,0.3 mmol)和乙腈(2 ml),80 o
c搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(20 mg,45 %)。ms: m/z [m+h]
+ =252。
[0112] 中间体21-苄基-5-氧吡咯烷-2-羧酸甲酯室温下,将5-氧吡咯烷-2-羧酸甲酯(4.0 g,28.0 mmol)和氢化钠(60 %,2.24 g,56.0 mmol)加入无水四氢呋喃(50 ml)中,氩气保护下升温至60 o
c,加入溴化苄(5.7 g,
33.6 mmol),60 o
c搅拌2小时。反应液冷却至室温后倒饱和氯化铵水溶液中,乙酸乙酯萃取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 3/1)得到标题化合物(4.2 g,64 %)。ms: m/z [m+h]
+ =234。
[0113]
1-苄基-5-(羟甲基)吡咯烷-2-酮室温下,将1-苄基-5-氧吡咯烷-2-羧酸甲酯(4.2 g,18.02 mmol)加入无水四氢呋喃(40ml)中,搅拌下加入硼氢化锂(800 mg,36.0 mmol),室温搅拌1小时。反应液用十水硫酸钠淬灭,室温搅拌30分钟。过滤,滤液浓缩得到标题化合物粗品(3.6 g,97 %)。ms: m/z [m+h]
+ =206。
[0114]
1-苄基-5-(((叔丁基二甲基硅烷基)氧基)甲基)吡咯烷-2-酮室温下,将1-苄基-5-(羟甲基)吡咯烷-2-酮(3.6 g,14.4 mmol)溶于二氯甲烷(50 ml)中,加入咪唑(2.9 g,43.2 mmol)和叔丁基二甲基氯硅烷(3.2 g,21.6 mmol),室温搅拌30分钟。反应液倒入饱和氯化铵水溶液中,二氯甲烷提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10/1)得到标题化合物(3.6 g,78 %)。ms: m/z [m+h]
+ =320。
[0115]
4-苄基5-(((叔丁基二甲基硅烷基)氧基)甲基)-4-氮杂螺[2. 4]庚烷室温下,将1-苄基-5-(((叔丁基二甲基硅烷基)氧基)甲基)吡咯烷-2-酮(2.0 g,6.27 mmol)加入无水四氢呋喃(30 ml)中,氩气保护下降温至0 o
c,搅拌下加入钛酸四异丙酯(4.5 ml,15.67 mmol)和乙基溴化镁的四氢呋喃溶液(2 m,15 ml,30 mmol),室温过夜。反应液用水淬灭,反应液过滤,滤液浓缩,柱层析纯化(石油醚/乙酸乙酯 = 20/1)得到标题化合物(1.3 g,63 %)。ms: m/z [m+h]
+ =332。
[0116] (4-苄基-4-氮杂螺[2. 4]庚烷-5-基)甲醇oc搅拌过夜。反应液过滤,滤液浓缩, 制备薄层色谱分离(二氯甲烷/甲醇 = 20/1)得到标题化合物(80 mg, 65 %)。 ms: m/z [m+h]
+ =238。
[0120] 中间体31, 1-双(碘甲基)环丙烷室温下,将三苯基膦(26.0 g,99.0 mmol)和咪唑(6.5 g,96.0 mmol)加入二氯甲烷(30 ml)中,氩气保护下降温至0 o
c,加入碘(25.0 g,99.0 mmol),0 o
c搅拌1小时。将1, 1-环丙烷-二甲醇(5.0 g,48.0 mmol)的二氯甲烷(20 ml)溶液加入反应液,该温度下搅拌3小时。反应液倒入食盐水(15 %)中,乙酸乙酯提取,有机相用饱和亚硫酸钠溶液洗涤,有机相浓缩,向残留物中依次加入石油醚(200 ml)和乙酸乙酯(10 ml),室温搅拌30分钟,过滤,滤液浓缩得到标题化合物(12 g,75 %)。1h nmr(400 mhz, cdcl3) d 3.34(br. s., 4 h), 1.02(br. s., 4 h)。
[0121] 5-(叔丁基)6-乙基 5-氮杂螺[2. 4]庚烷-5, 6-二羧酸酯室温下,将氢化钠(60 %,3.0 g,75.0 mmol)加入n, n-二甲基甲酰胺(40 ml)中,氩气保护下降温至0 o
c,搅拌下滴入1, 1-双(碘甲基)环丙烷(9.6 g,28.9 mmol)和(叔丁氧羰基)甘氨酸乙酯(6.2 g,30.5 mmol)的n, n-二甲基甲酰胺(40 ml)溶液,0 o
c搅拌1.5小时。加入乙酸(2.5 ml),0 o
c搅拌2小时。反应液倒入水中,乙酸乙酯提取,有机相浓缩得到标题化合物(4.8 g,62 %)。ms: m/z [m+h-boc]
+ =170。
[0122] (5-氮杂螺[2. 4]庚烷-6-基)甲醇盐酸盐室温下,将5-(叔丁基)6-乙基 5-氮杂螺[2. 4]庚烷-5, 6-二羧酸酯(540 mg,2.0 mmol)加入无水四氢呋喃(6 ml)中,搅拌下加入硼氢化锂(88 mg,4.0 mmol),室温搅拌过夜。反应液用十水硫酸钠淬灭,室温搅拌0.5小时,过滤,滤液浓缩,将残留物加入二氯甲烷(3 ml)和氯化氢的乙酸乙酯溶液(4 m,6 ml)中。室温搅拌30分钟,反应液浓缩得到标题化合物粗品(350 mg,107 %)。ms: m/z [m+h]
+ =128。
[0123] (5-(2, 6-二氯嘧啶-4-基)-5-氮杂螺[2. 4]庚烷-6-基)甲醇
室温下,将2, 4, 6-三氯嘧啶(760 mg,4.18 mmol)加入乙腈(12 ml)中,氩气保护下冷却至0 o
c,搅拌下依次加入碳酸钠固体(680 mg,6.42 mmol)和(5-氮杂螺[2. 4]庚烷-6-基)甲醇盐酸盐(350 mg,2.14 mmol),室温搅拌1小时。反应液过滤,滤液浓缩,制备薄层色谱分离(石油醚/乙酸乙酯 = 4/1)得到标题化合物(140 mg,19 %)。ms: m/z [m+h]
+ =273。
[0124] 3'-氯-8a', 9'-二氢-1'h, 6'h, 8'h-螺[环丙烷-1, 7'-吡咯并[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将(5-(2, 6-二氯嘧啶-4-基)-5-氮杂螺[2. 4]庚烷-6-基)甲醇(140 mg,0.51 mmol)加入二氯甲烷(6 ml)中,搅拌下滴加氯化亚砜(184 mg,1.53 mmol),室温搅拌10分钟。反应液浓缩,向残留物中依次加入碳酸钾固体(345 mg,2.5 mmol)和乙腈(8 ml),85 o
c搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离(二氯甲烷/甲醇 = 20/1)得到标题化合物(80 mg, 66 %)。ms: m/z [m+h] + =238。
[0125] 中间体45-羟基-6-氧杂-7-氮杂螺[3. 5]壬基-7-烯-8-羧酸乙酯室温下,将环丁烷甲醛(1 g,11.9 mmol)和吡咯烷(1.1 g,15.5 mmol)加到无水甲苯(12 ml)中,搅拌下加入4a分子筛(0.1 g),室温搅拌2小时。将无水四氢呋喃(12 ml)加入反应液,随后分批加入3-溴-2-(羟基亚氨基)丙酸乙酯(2.6 g,12.6 mmol,j. org. chem.,1982,47,2147),室温搅拌0.5小时。将三乙胺(1.3 g,12.7 mmol)加入反应液,室温搅拌过夜。反应液浓缩,柱层析纯化(石油醚/乙酸乙酯 = 5/1),制备薄层色谱纯化(石油醚/乙酸乙酯 = 5/1)得到标题化合物(510 mg,20 %)。1h nmr(400 mhz, dmso-d6)d 7.44
ꢀ‑ꢀ
7.14(m, 1 h), 5.26(br. s., 1 h), 4.21(q, j = 6.8 hz, 2 h), 2.56(m, 1 h), 2.32(m, 1 h), 2.09
ꢀ‑ꢀ
1.95(m, 1 h), 1.95
ꢀ‑ꢀ
1.66(m, 4 h), 1.59(d, j = 10.3 hz, 1 h), 1.25(t, j = 7.1 hz, 3 h); ms: m/z [m+h]
+ =214。
[0126] 6-氮杂螺[3. 4]辛烷-7-羧酸乙酯
室温下,将5-羟基-6-氧杂-7-氮杂螺[3. 5]壬基-7-烯-8-羧酸乙酯(510 mg,2.4 mmol)加入乙醇(30 ml)中,氩气保护下加入raney-ni催化剂(510 mg),室温常压氢化过夜。过滤,滤液浓缩得到标题化合物粗品(460 mg,102 %)。1h nmr(400 mhz, dmso-d6)d 4.06(d, j = 6.4 hz, 2 h), 3.63(br. s., 1 h), 2.86(br. s., 1 h), 2.76(br. s., 1 h), 2.04(d, j = 8.3 hz, 1 h), 1.96
ꢀ‑ꢀ
1.67(m, 7 h), 1.23
ꢀ‑ꢀ
1.11(m, 3 h); ms: m/z [m+h]
+ =184。
[0127]
(6-氮杂螺[3. 4]辛基-7-基)甲醇室温下,将6-氮杂螺[3. 4]辛烷-7-羧酸乙酯粗品(360 mg,1.97 mmol)加到无水四氢呋喃(10 ml)和甲醇(1 ml)中,缓慢加入硼氢化锂(220 mg,5.8 mmol),50 o
c搅拌2小时。反应液冷却到0 o
c,反应液用十水硫酸钠淬灭。过滤,滤饼用二氯甲烷淋洗,滤液浓缩得到标题化合物粗品(290 mg,105 %)。 ms: m/z [m+h]
+ =142。
[0128] (6-(6-氯-2-甲氧基嘧啶-4-基)-6-氮杂螺[3. 4]辛基-7-基)甲醇室温下,将(6-氮杂螺[3. 4]辛基-7-基)甲醇(290 mg,2.06 mmol)和4, 6-二氯-2-甲氧基嘧啶(358 mg,2 mmol)溶解在异丙醇(20 ml)中,搅拌下加入碳酸钠(848 mg,8 mmol),80 o
c搅拌3小时。反应液冷却到室温,用二氯甲烷稀释反应混合物,硅藻土过滤,滤液浓缩,制备薄层色谱分离(石油醚/乙酸乙酯 = 2/1)得到标题化合物(160 mg,24 %)。1h nmr(400 mhz, cdcl3)d 6.05(br. s., 1 h), 5.55(br. s., 1 h), 4.29(br. s., 1 h), 3.93(s, 3 h), 3.78
ꢀ‑ꢀ
3.60(m, 2 h), 3.53
ꢀ‑ꢀ
3.42(m, 1 h), 3.39(br. s., 1 h), 2.17
ꢀ‑ꢀ
2.10(m, 1 h), 2.10
ꢀ‑ꢀ
1.85(m, 7 h); ms: m/z [m+h]
+ =284。
[0129] 3'-氯-8a', 9'-二氢-1'h, 6'h, 8'h-螺[环丁烷-1, 7'-吡咯并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮
室温下,将(6-(6-氯-2-甲氧基嘧啶-4-基)-6-氮杂螺[3. 4]辛基-7-基)甲醇(140 mg, 0.49 mmol)和氯化亚砜(176 mg,1.48 mmol)加入到二氯甲烷(3 ml)中,室温搅拌过夜。反应液浓缩并溶解在水(2 ml)中,0 o
c下加入氢氧化钠固体(78 mg,1.96 mmol),0 o
c搅拌0.5小时。反应液倒入水中,用二氯甲烷萃取,有机相浓缩,制备薄层色谱分离(二氯甲烷/甲醇 = 40/1)得到白色固体标题化合物(47 mg,38 %)。1h nmr(400 mhz, cdcl3)d 5.60(s, 1 h), 4.22(d, j = 7.8 hz, 2 h), 4.09
ꢀ‑ꢀ
3.97(m, 1 h), 3.47(d, j = 10.8 hz, 1 h), 3.23(d, j = 10.8 hz, 1 h), 2.37
ꢀ‑ꢀ
2.27(m, 1 h), 2.18
ꢀ‑ꢀ
1.88(m, 7 h); ms: m/z [m+h]
+ =252。
[0130] 中间体52-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-甲烯基哌啶-1-羧酸叔丁酯室温下,将甲基三苯基溴化磷(407 mg,1.14 mmol)加入无水四氢呋喃(10 ml)中,氩气保护下降温至0 o
c,滴加正丁基锂的正己烷溶液(2.5 m,0.456 ml,1.14 mml),0 o
c搅拌0.5小时。将2-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-氧代哌啶-1-羧酸叔丁酯(300 mg,0.87 mmol,bioorganic and medicinal chemistry letters, 2017, 27, 2210
ꢀ‑ꢀ
2215)的无水四氢呋喃(10 ml)溶液缓慢滴入反应液中,室温搅拌过夜。反应液冷却到0 o
c,加入饱和氯化铵溶液淬灭,乙酸乙酯萃取,有机相浓缩,制备薄层色谱分离(二氯甲烷/甲醇 = 100/1)得到标题化合物(92 mg,29 %)。ms: m/z [m+h-boc]
+ =242。
[0131] (5-氮杂螺[2. 5]辛基-4-基)甲醇盐酸盐室温下,将2-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-甲烯基哌啶-1-羧酸叔丁酯(82 mg,0.24 mmol)和二乙基锌的正己烷溶液(1 m,2.4 ml,2.4 mml)加入二氯甲烷(20 ml)中,氩气保护下冷却到0 o
c,滴加二碘甲烷(1.3 g,4.8 mmol),滴加完后室温搅拌过夜。反应液冷却到0 o
c,反应液用十水硫酸钠淬灭,过滤,滤饼用二氯甲烷淋洗,滤液浓缩,向残留物中依次加入甲醇(2 ml)和氯化氢的乙酸乙酯溶液(4 m,1 ml),室温搅拌5小时。反应液浓缩得到标题化合物粗品(90 mg,211 %)。ms: m/z [m+h]
+ =142。
[0132] (5-(6-氯-2-甲氧基嘧啶-4-基)-5-氮杂螺[2. 5]辛基-4-基)甲醇室温下,将(5-氮杂螺[2. 5]辛基-4-基)甲醇盐酸盐粗品(90 mg,0.51 mmol)、4,6-二氯-2-甲氧基嘧啶(90 mg,0.39 mmol)和碳酸钠固体(127 mg,1.2 mmol)加入异丙醇(5 ml)中,80 o
c搅拌2天。反应液冷却到室温后倒入水中,二氯甲烷提取,有机相浓缩,制备薄层色谱分离(石油醚/乙酸乙酯 = 2/1)得到标题化合物(20 mg,14 %)。1h nmr(400 mhz, cdcl3)d 6.24(s, 1 h), 4.79
ꢀ‑ꢀ
4.50(m, 2 h), 4.01(br. s., 1 h), 3.88(s, 3 h), 3.81(d, j = 7.3 hz, 1 h), 3.14(d, j = 7.3 hz, 1 h), 2.19
ꢀ‑ꢀ
1.93(m, 2 h), 1.74(br. s., 1 h), 1.71
ꢀ‑ꢀ
1.59(m, 1 h), 0.88
ꢀ‑ꢀ
0.76(m, 1 h), 0.68
ꢀ‑ꢀ
0.49(m, 1 h), 0.47
ꢀ‑ꢀ
0.36(m, 1 h), 0.21(br. s., 1 h); ms: m/z [m+h]
+ =284。
[0133] 3'-氯-7', 8', 9a', 10'-四氢-1'h, 6'h-螺环[1, 9'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮室温下,将(5-(6-氯-2-甲氧基嘧啶-4-基)-5-氮杂螺[2. 5]辛基-4-基)甲醇(20 mg,0.07 mmol)和氯化亚砜(26 mg,0.22 mmol)加入到二氯甲烷(3 ml)中,室温搅拌过夜。反应液浓缩,向残留物中加入水(2 ml),冷却至0 o
c,搅拌下加入氢氧化钠固体(11 mg,0.28 mmol)。0 o
c搅拌0.5小时。反应液倒入水中,二氯甲烷萃取,有机相浓缩得到标题化合物粗品(24 mg,113 %)。ms: m/z [m+h]
+ =252。
[0134] 中间体63-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯室温下,将3-氮杂螺[5. 5]十一烷(900 mg,5.87 mmol)、二碳酸二叔丁酯(1.92 g,8.81 mmol)和碳酸钠(1.87 g,17.61 mmol)依次加入二氯甲烷(10ml)中,室温下搅拌5小时。反应液倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(1.62 g,94 %)。ms: m/z [m+h-tbu]
+ =198。
[0135] 2-甲酰基-3-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯
氩气保护下,将3-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯(1 g,3.95 mmol)和四甲基乙二胺(458 mg,3.95 mmol)加入无水四氢呋喃(10 ml)中,氩气保护下降温至-60 o
c,滴加仲丁基锂的正己烷溶液(1.3 m,3.03 ml,3.95 mmol),
ꢀ‑
50 o
c ~
ꢀ‑
20 o
c搅拌0.5小时。反应液降温至-60 o
c,滴加n,n-二甲基甲酰胺(432 mg,5.93 mmol),-50 o
c搅拌1小时,室温搅拌1小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(563 mg,51 %)。ms: m/z [m+h-tbu]
+ =226。
[0136] 2-(羟甲基)-3-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯室温下,将2-甲酰基-3-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯(460 mg,1.64 mmol)加入乙醇(5 ml)中,搅拌下加入硼氢化钠(124 mg,3.27 mmol),室温搅拌2小时。反应液用饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩得到标题化合物(430 mg,93 %)。ms: m/z [m+h-tbu]
+ =228。
[0137] (3-(2, 6-二氯嘧啶-4-基)-3-氮杂螺环[5. 5]十一烷-2-基)甲醇室温下,将2-(羟甲基)-3-氮杂螺[5. 5]十一烷-3-羧酸叔丁酯(430 mg,1.52 mmol)加入二氯甲烷(3 ml)中,搅拌下加入氯化氢的乙酸乙酯溶液(4 m,1.9 ml),室温搅拌2小时。反应液浓缩,向残留物中依次加入乙腈(5 ml)和碳酸钠(483 mg,4.56 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱分离得到标题化合物(410 mg,8 2%)。ms: m/z [m+h]
+ =330。
[0138] 3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺[环己烷-1, 8'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮
室温下,将(3-(2, 6-二氯嘧啶-4-基)-3-氮杂螺环[5. 5]十一烷-2-基)甲醇(360 mg,1.09 mmol)加入二氯甲烷(5 ml)中,搅拌下加入氯化亚砜(389 mg,3.27 mmol),室温搅拌1小时。反应液浓缩,向残留物中依次加入乙腈(5 ml)和碳酸钾(451 mg,3.1 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱纯化得到标题化合物(220 mg,60 %)。 ms: m/z [m+h]
+ =294。
[0139] 中间体71, 1-二氯-2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,将4-亚甲基哌啶-1-羧酸叔丁酯(49 g,248.4 mmol)和锌铜试剂(49 g)依次加入无水乙二醇二甲醚(500 ml)中,控温30 o
c-40 o
c,滴加三氯乙酰氯(49.4 g,273.2 mmol),室温搅拌3小时。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,柱层析纯化得到标题化合物(38.6 g,50 %)。ms: m/z [m+h-tbu]
+ =252。
[0140] 2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,依次将1, 1-二氯-2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(37 g,120.1 mmol)加入饱和氯化铵水溶液(200 ml)和甲醇(200 ml)中,氩气保护下降温至0 o
c,分批加入锌粉(37 g,565.8 mmol),室温搅拌过夜。反应液过滤,滤饼用乙酸乙酯淋洗,有机相浓缩,柱层析纯化得到标题化合物(24.2 g,84 %)。 ms: m/z [m+h-tbu]
+ =184。
[0141] 7-氮杂螺[3. 5]壬烷室温下,将2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(5 g,20.92 mmol)加入聚乙二醇(50 ml)中,搅拌下加入水合肼(7.32 g,146.44 mmol)和氢氧化钾固体(7.03 g,
125.52 mmol),200 o
c搅拌4小时。反应液冷却至室温后倒入水中,乙醚提取,有机相浓缩得到标题化合物(3.32 g,126 %)。ms: m/z [m+h]
+ =126。
[0142] 7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯室温下,将7-氮杂螺[3. 5]壬烷(2.8 g,22.4 mmol)加入二氯甲烷(50 ml)中,搅拌下依次加入二碳酸二叔丁酯(7.32 g,33.6 mmol)和碳酸钾(9.27 g,67.3 mmol),室温下搅拌过夜。反应液倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(4.5 g,89 %)。ms: m/z [m+h-tbu]
+ =170。
[0143] 6-甲酰基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯室温下,依次将四甲基乙二胺(1.03 g,8.89 mmol)和7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(2 g,8.89 mmol)加入无水四氢呋喃(20 ml)中,氩气保护下降温至-60 o
c,搅拌下滴加仲丁基锂的正己烷溶液(1.3 m,6.84 ml,8.89 mmol),滴加结束-20 o
c搅拌30分钟。反应液降温至-60 o
c,滴加n,n-二甲基甲酰胺(973 mg,13.3 mmol),滴加结束-60 o
c搅拌1小时,室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(0.83 g,37 %)。ms: m/z [m+h-tbu]
+ =198。
[0144]
6-(羟甲基)-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯室温下,将6-甲酰基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(1.63 g,6.4 mmol)加入无水乙醇(10 ml)中,搅拌下加入硼氢化钠(487 mg,12.8 mmol),室温搅拌2小时。反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩得到标题化合物(1.43 g,87 %)。ms: m/z [m+h-tbu]
+ =200。
[0145] (7-(6-氯-2-甲氧基嘧啶-4-基)-7-氮杂螺[3. 5]壬烷-6-基)甲醇
室温下,将6-(羟甲基)-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(1.43 g,6.4 mmol)加入二氯甲烷(100 ml)中,搅拌下加入氯化氢的乙酸乙酯溶液(4 m,27.8 ml),室温搅拌1小时。反应液浓缩,向残留物中依次加入乙腈(10 ml)、4,6-二氯-2-甲氧基嘧啶(2.29 g,12.8 mmol)和碳酸钠(2.32 g, 21.87 mmol),85 o
c搅拌16小时。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,柱层析纯化得到标题化合物(0.783 g,35 %)。ms: m/z [m+h]
+ =298。
[0146] 3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将(7-(6-氯-2-甲氧基嘧啶-4-基)-7-氮杂螺[3. 5]壬烷-6-基)甲醇(100 mg,0.33 mmol)加入二氯甲烷(5 ml)中,搅拌下加入氯化亚砜(118 mg,0.99 mmol),室温搅拌1小时。反应液浓缩,向残留物中依次加入乙腈(5 ml)和碳酸钾(182 mg,1.32 mmol), 85 o
c搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱纯化得到标题化合物(38 mg,43 %)。 ms: m/z [m+h]
+ =266。
[0147] 中间体88-氮杂螺环[4. 5]癸烷-8-羧酸叔丁酯室温下,将8-氮杂螺[4. 5]癸烷盐酸盐(1 g,7.18 mmol)加入二氯甲烷(10 ml)中,搅拌下依次加入二碳酸二叔丁酯(2.35 g,10.77 mmol)和碳酸钾(2.98 g,21.54 mmol),室温搅拌过夜。反应液倒入水中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(1.41 g,82 %)。ms: m/z [m+h-tbu]
+ =184。
[0148] 7-甲酰基-8-氮杂螺环[4. 5]癸烷-8-羧酸叔丁酯
ms: m/z [m+h]
+ =280。
[0152] 中间体82-羟基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,将2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(5 g,20.89 mmol)加入甲醇(50ml)中,搅拌下加入硼氢化钠(2.37 g,62.67 mmol),室温搅拌5小时。反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩得到标题化合物(4.93 g,98 %)。 ms: m/z [m+h-tbu]
+ =186。
[0153] 2-((叔丁基二甲基甲硅烷基)氧基)-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯室温下,依次将2-羟基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(4.83 g,20.01 mmol)和咪唑(2.72 g,40.02 mmol)加入二氯甲烷(50 ml)中,搅拌下加入叔丁基二甲基氯硅烷(4.52 g,30.01 mmol),室温搅拌3小时。反应液倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(6.8 g,96 %)。 ms: m/z [m+h-tbu]
+ =300。
[0154] 2-((叔丁基二甲基硅基)氧基)-6-甲酰基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,依次将四甲基乙二胺(0.69 g,5.91 mmol)和2-((叔丁基二甲基甲硅烷基)氧基)-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(2.1 g,5.91 mmol)加入无水四氢呋喃(15 ml)中,氩气保护下降温至-60 o
c,搅拌下滴加仲丁基锂的正己烷溶液(1.3 m,4.55 ml,5.91 mmol),滴加结束-20 o
c搅拌30分钟。反应液降温至-60 o
c,滴加n,n-二甲基甲酰胺(650 mg,8.87 mmol),滴加结束-60 o
c搅拌1小时,室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(2.2 g,97 %)。 ms: m/z [m+h-tbu]
+ =328。
[0155] 2-(叔丁基二甲基硅基)氧基)-6-(羟甲基)-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,将2-((叔丁基二甲基硅基)氧基)-6-甲酰基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(310 mg,0.81 mmol)加入无水甲醇(3 ml)中,搅拌下加入硼氢化钠(91 mg,2.43 mmol),室温搅拌3小时,反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩得到标题化合物(298 mg,95 %)。ms: m/z [m+h-tbu]
+ =330。
[0156] (2-((叔丁基二甲基硅基)氧基)-7-(6-氯-2-甲氧基嘧啶-4-基)-7-氮杂螺环[3. 5]壬烷-6-基)甲醇室温下,将2-(叔丁基二甲基硅基)氧基)-6-(羟甲基)-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(296 mg,0.77 mmol)加入二氯甲烷(10 ml)和三氟乙酸(1 ml)中,室温搅拌1小时。加入碳酸钾(320 mg,2.31 mmol),室温搅拌0.5小时。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩。向残留物中依次加入乙腈(5 ml)、4,6-二氯-2-甲氧基嘧啶(196 mg,1.06 mmol)和碳酸钠(169 mg,1.59 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱分离得到标题化合物(55 mg,17 %)。ms: m/z [m+h]
+ =428。
[0157] 3-((叔丁基二甲基硅基)氧基)-3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将(2-((叔丁基二甲基硅基)氧基)-7-(6-氯-2-甲氧基嘧啶-4-基)-7-氮杂螺环[3. 5]壬-6-基)甲醇(55 mg,0.13 mmol)和三乙胺(66 mg,0.26 mmol)加入二氯甲烷(5 ml)中,搅拌下加入甲基磺酸酐(45 mg,0.26 mmol),室温搅拌3小时。反应液倒入水中,二氯甲烷提取,有机相浓缩,向残留物中依次加入水(5 ml)和氢氧化钠水溶液(15 %,123 mg), 室温搅拌0.5小时。反应液用二氯甲烷提取,有机相浓缩得到标题化合物(18 mg,
41 %)。ms: m/z [m+h]
+ =396。
[0158] 3-((叔丁基二甲基硅基)氧基)-3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮将3-((叔丁基二甲基硅基)氧基)-3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮(17 mg,0.043 mmol)、(3-氟
ꢀ‑
4-(2-三氟甲基)吡啶-4-基)苯基)甲醇(37 mg,0.13 mmol)和碳酸铯(42 mg,0.13 mmol)加入甲苯(5 ml)中,110 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱纯化得到标题化合物(23 mg,83 %)。ms: m/z [m+h]
+ =647。
[0159] 中间体92-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,将2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(5 g,20.89 mmol)加入甲醇(50 ml)中,搅拌下加入硼氢化钠(2.37 g,62.67 mmol),室温搅拌5小时。反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩,向残留物中依次加入乙腈(30 ml)和碘甲烷(8.96 g,62.67 mmol),搅拌下加入氢化钠(60 %,2.51 g,62.67 mmol),室温搅拌3小时。反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(1.9 g,36 %)。ms: m/z [m+h-tbu]
+ =200。
[0160] 6-甲酰基-2-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,依次将四甲基乙二胺(1.3 g,11.16 mmol)和2-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(1.9 g,7.44 mmol)加入无水四氢呋喃(20 ml)中,氩气保护下降温至-60 o
c,搅拌下滴加仲丁基锂的正己烷溶液(1.3 m,8.58 ml,11.16 mmol),滴加结束-20 o
c搅拌30分钟。反应液降温至-60 o
c,滴加n, n-二甲基甲酰胺(1.63 g,22.32 mmol),滴加
结束-60 o
c搅拌1小时,室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(0.57 g,27 %)。 ms: m/z [m+h-tbu]
+ =228。
[0161] 6-(羟甲基)-2-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯室温下,将6-甲酰基-2-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(380 mg,1.34 mmol)加入无水甲醇(5 ml)中,搅拌下加入硼氢化钠(150 mg,4.02 mmol),室温搅拌3小时,反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩得到标题化合物(365 mg,95 %)。ms: m/z [m+h-tbu]
+ =230。
[0162] (7-(6-氯-2-甲氧基嘧啶-4-基)-2-甲氧基-7-氮杂螺环[3. 5]壬烷-6-基)甲醇室温下,将6-(羟甲基)-2-甲氧基-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(365 mg,1.28 mmol)加入二氯甲烷(2 ml)中,搅拌下加入氯化氢的乙酸乙酯溶液(4 m,2 ml),室温搅拌1小时。反应液浓缩,向残留物中依次加入乙腈(10 ml)、4,6-二氯-2-甲氧基嘧啶(344 mg,1.92 mmol)和碳酸钠(385 mg,3.63 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱分离得到标题化合物(310 mg,74 %)。ms: m/z [m+h]
+ =328。
[0163] 3'-氯-3-甲氧基-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将(7-(6-氯-2-甲氧基嘧啶-4-基)-2-甲氧基-7-氮杂螺环[3. 5]壬烷-6-基)甲醇(270 mg,0.82 mmol)加入二氯甲烷(5 ml)中,搅拌下加入氯化亚砜(118 mg,0.99 mmol),室温搅拌1小时。反应液浓缩,向残留物中依次加入乙腈(5 ml)和碳酸钾固体(298 mg,2.16 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱纯化得到标题化合物(198 mg,93 %)。 ms: m/z [m+h]
+ =296。
[0164] 中间体101, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯室温下,将2-氧代-7-氮杂螺环[3. 5]壬烷-7-羧酸叔丁酯(5 g,20.89 mmol)、乙二醇(1.94 g,31.34 mmol)和4-甲基苯磺酸吡啶(1.05 g,4.18 mmol)依次加入甲苯(50 ml)中, 120 o
c搅拌过夜。反应液浓缩,柱层析纯化得到标题化合物(3.6 g,61 %)。 ms: m/z [m+h-tbu]
+ =228。
[0165] 9-甲酰基-1, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯室温下,依次将四甲基乙二胺(2.15 g,18.52 mmol)和1, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯(3.5 g,12.35 mmol)加入无水四氢呋喃(40 ml)中,氩气保护下降温至-60 o
c,搅拌下滴加仲丁基锂的正己烷溶液(1.3 m,14.25 ml,18.52 mmol),滴加结束-20 o
c搅拌30分钟。反应液降温至-60 o
c,滴加n, n-二甲基甲酰胺(2.71 g,37.05 mmol),滴加结束-60 o
c搅拌1小时,室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(1.23 g,32 %)。ms: m/z [m+h-tbu]
+ =256。
[0166] 9-(羟甲基)-1, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯室温下,将9-甲酰基-1, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯(1.13 g,3.63 mmol)加入无水甲醇(10 ml)中,搅拌下加入硼氢化钠(270 mg,7.26 mmol),室温搅拌3小时,反应液用饱和氯化铵水溶液淬灭,二氯甲烷提取,有机相浓缩得到标题化合物(1.1 g,97 %)。ms: m/z [m+h-tbu]
+ =258。
[0167]
7-(6-氯-2-甲氧基嘧啶-4-基)-6-(羟甲基)-7-氮杂螺环[3. 5]壬-2-酮
室温下,将9-(羟甲基)-1, 4-二氧杂-10-氮杂双螺[4. 1. 57. 15]十三烷-10-羧酸叔丁酯(1.1 g,3.51 mmol)加入二氯甲烷(10 ml)中,搅拌下加入氯化氢的乙酸乙酯溶液(4 m,0.32 ml),室温搅拌3小时。反应液浓缩,向残留物中依次加入乙腈(10 ml)、4, 6-二氯-2-甲氧基嘧啶(942 mg,5.27 mmol)和碳酸钠(2.07 g ,19.5 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱分离得到标题化合物(180 mg,8%)。ms: m/z [m+h]
+ =312。
[0168] 3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1', 3-二酮室温下,将7-(6-氯-2-甲氧基嘧啶-4-基)-6-(羟甲基)-7-氮杂螺环[3. 5]壬烷-2-酮(58 mg,0.19 mmol)加入二氯甲烷(5 ml)中,搅拌下加入氯化亚砜(67.8 mg,0.57 mmol),室温搅拌3小时。反应液浓缩,向残留物中依次加入乙腈(3 ml)和碳酸钾固体(62 mg,0.45 mmol),85 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱纯化得到标题化合物(37 mg,88 %)。ms: m/z [m+h]
+ =280。
[0169] 3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1', 3-二酮室温下,依次将3'-氯-3-甲氧基-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮(33 mg,0.12 mmol)、(3-氟-4-(2-三氟甲基)吡啶-4-基)苯基)甲醇(51 mg,0.18 mmol)和碳酸铯(78 mg,0.24 mmol)加入甲苯(3 ml)中,120 o
c搅拌过夜。反应液过滤,滤饼用二氯甲烷淋洗,滤液浓缩,制备薄层色谱纯化得到标题化合物(41 mg,64 %)。ms: m/z [m+h]
+ =531。
[0170] 中间体112-甲氧基-6-乙烯基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯
室温下,将6-甲酰基-2-甲氧基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(2.4 g,8.47 mmol)加入无水四氢呋喃中(80 ml)中,搅拌下加入甲基三苯基溴化磷(3.63 g,10.16 mmol),氩气保护下降温至0 o
c,分批次加入氢化钠(60%,1.02 g,25.41 mmol),加料结束室温搅拌3小时。反应液用饱和氯化铵水溶液淬灭,乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10/1)得到标题化合物(700 mg,29 %)。ms: m/z [m+h-tbu]
+ =226。
[0171] 6-(2-羟乙基)-2-甲氧基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯室温下,将2-甲氧基-6-乙烯基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(700 mg,2.49 mmol)加入9-硼双环[3. 3. 1]壬烷的四氢呋喃溶液(0.5 m,20 ml,9.96 mmol)中,室温搅拌过夜。向反应液中依次加入水(2 ml)、氢氧化钠水溶液(3 m,10 ml)和双氧水(37 %,10 ml),反应液50 o
c搅拌2小时。将反应液降温至室温后用乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 2/1)得到标题化合物(750 mg,100 %)。ms: m/z [m+h-tbu]
+ =244。
[0172] 2-(2-甲氧基-7-氮杂螺[3. 5]壬烷-6-基)乙基-1-醇盐酸盐室温下,将6-(2-羟乙基)-2-甲氧基-7-氮杂螺[3. 5]壬烷-7-羧酸叔丁酯(750 mg,2.50 mmol)加入氯化氢的1,4-二氧六环溶液(10 ml)中,室温搅拌2小时。反应液浓缩得到标题化合物粗品(590 mg,100 %)。ms: m/z [m+h]
+ =200。
[0173] 2-(7-(2, 6-二氯嘧啶-4-基)-2-甲氧基-7-氮杂螺[3. 5]壬烷-6-基)乙基-1-醇
室温下,将2-(2-甲氧基-7-氮杂螺[3. 5]壬烷-6-基)乙基-1-醇盐酸盐(570 mg,2.86 mmol)加入乙腈(10 ml)中,搅拌下依次加入4, 6-二氯-2-甲氧基嘧啶(512 mg,2.86 mmol)和碳酸钠固体(909 mg,8.58 mmol),反应液室温搅拌过夜。反应液倒入水中,乙酸乙酯提取,有机相浓缩,柱层析纯化(二氯甲烷/甲醇 = 50/1)得到标题化合物(390 mg,40 %)。ms: m/z [m+h]
+ =342。
[0174] 7-(6-氯-2-甲氧基嘧啶-4-基)-6-(2-氯乙基)-2-甲氧基-7-氮杂螺[3. 5]壬烷室温下,将2-(7-(2, 6-二氯嘧啶-4-基)-2-甲氧基-7-氮杂螺[3. 5]壬烷-6-基)乙-1-醇(350 mg,1.02 mmol)加入二氯甲烷(20 ml)中,降温至0 o
c,加入氯化亚砜(394 mg,3.06 mmol),回流搅拌2小时。反应液浓缩,得到标题化合物(350 mg,95%)。ms: m/z [m+h]
+ =360。
[0175]
2'-氯-3-甲氧基-6', 7', 7a', 8', 10', 11'-六氢-4'h-螺[环丁烷-1, 9'-吡啶并[1, 2-c] 嘧啶并[1, 6-a]嘧啶]-4'-酮氩气保护下,将7-(6-氯-2-甲氧基嘧啶-4-基)-6-(2-氯乙基)-2-甲氧基-7-氮杂螺[3. 5]壬烷(350 mg,0.97 mmol)和碳酸钾(402 mg,2.91 mmol)加入乙腈(10 ml)中,85oc搅拌过夜,反应液过滤,有机相浓缩,柱层析纯化(二氯甲烷/甲醇 = 100/1~50/1)得到标题化合物(200 mg,66 %)。ms: m/z [m+h]
+ =310。
[0176] 中间体126-氧哌啶-2-羧酸
室温下,将6-氧代-1, 6-二氢吡啶-2-羧酸(10 g,72 mmol)和钯碳(10%,1 g)加入甲醇(100 ml)中,室温常压氢化过夜。反应液过滤,滤液浓缩得到标题化合物(5 g,50%)。lc-ms: m/z [m+h]
+ =144。
[0177] 6-氧哌啶甲基-2-羧酸甲酯氩气保护下,将甲醇(10 ml)冷却至0 o
c,搅拌下滴入二氯亚砜(830 mg,7.7 mmol),室温搅拌10分钟。将6-氧哌啶-2-羧酸(1 g,7 mmol)加入反应液,室温搅拌过夜。反应液浓缩,向残留物中依次加入甲苯(10 ml)和三乙胺(1.4 g,14 mmol),室温搅拌0.5小时,反应液过滤,滤液浓缩得到标题化合物(850 mg,77 %)。ms: m/z [m+h]
+ =158。
[0178] 1-苄基-6-((苄氧基)甲基)哌啶-2-酮室温下,将6-氧哌啶甲基-2-羧酸甲酯(850 mg,5.4 mmol)加入甲醇(10 ml)中,搅拌下缓慢加入硼氢化钠(307 mg,8.1 mmol),室温搅拌1小时。反应液用十水硫酸钠淬灭,过滤,滤液浓缩。将残留物加入二甲亚砜(20 ml)中,氩气保护下降温至0 o
c,搅拌下缓慢加入氢化钠(3.6 g,21 mmol),室温搅拌30分钟。将溴化苄(3.6 g,21 mmol)加入反应液,室温搅拌过夜。反应液用水淬灭后倒入水中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(900 mg,54 %)。ms: m/z [m+h]
+ =310。
[0179] 4-苄基-5-((苄氧基)甲基)-4-氮杂螺[2. 5]辛烷氩气保护下,将无水四氢呋喃(60 ml)降温至-40 o
c,搅拌下依次加入乙基溴化镁的四氢呋喃溶液(1m,20 ml,19.4 mmol)和钛酸四异丙酯(2 g,7.2 mmol),室温搅拌5分钟。将1-苄基-6-((苄氧基)甲基)哌啶-2-酮(2.0 g,6.5 mmol)的无水四氢呋喃(1 ml)溶液加入反应液,回流搅拌过夜。反应液用10%氢氧化钠水溶液淬灭,无水硫酸钠干燥,过滤,滤液浓缩,柱层析纯化得到标题化合物(1.1 g,53 %)。ms: m/z [m+h]
+ =322。
[0180] (4-氮杂螺[2. 5]辛-5-基)甲醇盐酸盐
室温下,将4-苄基-5-((苄氧基)甲基)-4-氮杂螺[2. 5]辛烷(200 mg,0.6 mmol)和钯碳(10%,30 mg)加入氯化氢的乙酸乙酯溶液(4 m,5 ml)和甲醇(5 ml)中,40 o
c常压氢化过夜。反应液过滤,滤液浓缩得到标题化合物粗品(200 mg,182 %)。ms: m/z [m+h]
+ =142。
[0181] (4-(2, 6-二氯嘧啶-4-基)-4-氮杂螺[2. 5]辛基-5-基)甲醇室温下,依次将(4-氮杂螺[2. 5]辛烷-5-基)甲醇盐酸盐(100 mg,0.6 mmol)、2, 4, 6-三氯嘧啶(146 mg,0.8 mmol)和碳酸钠固体(149 mg,1.4 mmol)加入乙腈(2 ml)中,室温搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(100 mg,62 %)。ms: m/z [m+h]
+ =288。
[0182] 3'-氯-8', 9', 9a', 10'-四氢-1'h, 7'h-螺[环丙烷-1, 6'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮室温下,将(4-(2, 6-二氯嘧啶-4-基)-4-氮杂螺[2. 5]辛基-5-基)甲醇(100 mg,0.3 mmol)和二氯亚砜(47 mg,0.4 mmol)加入二氯甲烷(2 ml)中,室温搅拌过夜。反应液浓缩,向残留物中依次加入碳酸钾固体(83 mg,0.6 mmol)和乙腈(5 ml),80 o
c搅拌过夜。反应液冷却至室温,过滤,滤液浓缩,制备色谱分离得到标题化合物(80 mg,100 %)。ms: m/z [m+h]
+ =252。
[0183] 中间体134-亚甲基哌啶-1-羧酸苄酯室温下,将甲基三苯基溴化磷(10 g,27.8 mmol)加入无水四氢呋喃(30 ml)中,氩气保护下降温至0 o
c,控温0 o
c以下滴加正丁基锂的四氢呋喃溶液(2.5 m,11 ml,27.8 mmol),0 o
c搅拌1小时。控温0 o
c以下将4-氧哌啶-1-甲酸苄酯(5 g,21.4 mmol)的无水四氢呋喃(20 ml)溶液滴入反应液,0 o
c搅拌1小时后室温搅拌过夜。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(3.8 g,76 %)。ms: m/z [m+h]
+ =232。
[0184]
6-氮杂螺并[2. 5]辛烷-6-羧酸苄酯
氩气保护下,将二氯甲烷(60 ml)降温至0 o
c,搅拌下加入二乙基锌的正己烷溶液(1 m,60 ml,60 mmol),控温0 o
c以下滴入三氟乙酸(6.8 g,60 mmol),0 o
c搅拌0.5小时。将二碘甲烷(16 g,60 mmol)加入反应液,室温搅拌30分钟。将4-亚甲基哌啶-1-羧酸苄酯(7 g,30 mmol)加入反应液,室温反应过夜。反应液倒入饱和碳酸氢钠水溶液中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(6.1 g,82 %)。ms: m/z [m+h]
+ =246。
[0185] 6-氮杂螺[2. 5]辛烷盐酸盐室温下,将6-氮杂螺并[2. 5]辛烷-6-羧酸苄酯(8.3 g,33.9 mmol)和钯碳(10%,830 mg)加入甲醇(30 ml)中,室温常压氢化过夜。反应液过滤,向滤液中加入氯化氢的1, 4-二氧六环溶液(4 m,30 ml),室温搅拌30分钟,反应液浓缩,向残留物中加入四氢呋喃(15 ml),室温搅拌30分钟。过滤,将滤饼烘干得到标题化合物(4.3 g,86 %)。ms: m/z [m+h]
+ =112。
[0186] 6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯室温下,依次将6-氮杂螺[2. 5]辛烷盐酸盐(4.27 g,29 mmol)、三乙胺(6.2 g,61 mmol)和二碳酸二叔丁酯(7 g,32 mmol)加入二氯甲烷(50 ml)中,室温搅拌过夜。反应液倒入饱和氯化铵水溶液中,二氯甲烷提取,有机相浓缩,柱层析纯化得到标题化合物(5.3 g,87 %)。ms: m/z [m+h]
+ =212。
[0187] 5-甲酰基-6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯室温下,将6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯(400 mg,1.9 mmol)加入无水四氢呋喃(5 ml)中,氩气保护下降温至-78 o
c,控温-50 o
c以下依次滴入四甲基乙二胺(660 mg,5.7 mmol)和仲丁基锂的正己烷溶液(1.3 m,2.2 ml 2.9 mmol),-30 o
c搅拌15分钟。降温至-78 o
c,滴入n, n-二甲基甲酰胺(416 mg,5.7 mmol),室温搅拌过夜。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化得到标题化合物(110 mg,24 %)。ms: m/z [m+h]
+ =240。
[0188] 5-(羟甲基)-6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯室温下,将5-甲酰基-6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯(110 mg,0.46 mmol)加入无水乙醇(5 ml)中,搅拌下加入硼氢化钠固体(17 mg,0.46 mmol),室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩得到标题化合物(110 mg,100 %)。ms: m/z [m+h]
+ =242。
[0189] (6-(2, 6-二氯嘧啶-4-基)-6-氮杂螺[2. 5]辛基-5-基)甲醇
室温下,将5-(羟甲基)-6-氮杂螺[2. 5]辛烷-6-羧酸叔丁酯(360 mg,1.5 mmol)加入氯化氢的乙酸乙酯溶液(4 m,5 ml)和二氯甲烷(5 ml)中,室温搅拌1小时。反应液浓缩,向残留物中依次加入碳酸钠固体(318 mg,3.0 mmol)、2,4,6-三氯嘧啶(366 mg,3.0 mmol)和乙腈(5 ml),室温搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(260 mg,60 %)。ms: m/z [m+h]
+ =288。
[0190] 3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺[环丙烷-1, 8'-吡啶基[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,依次将(6-(2, 6-二氯嘧啶-4-基)-6-氮杂螺[2. 5]辛基-5-基)甲醇(160 mg,0.6 mmol)和二氯亚砜(1 ml)加入二氯甲烷(1 ml)中,室温搅拌过夜。反应液浓缩,向残留物中依次加入碳酸钾固体(230 mg,1.7 mmol)和乙腈(5 ml),回流搅拌过夜。反应液冷却至室温后过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(80 mg,50 %)。ms: m/z [m+h]
+ =252。
[0191]
中间体14 8-甲酰基-3-氧杂-9-氮杂螺[5. 5]十一烷-9-羧酸叔丁酯室温下,将3-氧杂-9-氮杂螺[5. 5]十一烷-9-羧酸叔丁酯(750 mg,2.94 mmol)和四甲基乙二胺(0.51 g,4.41 mmol)加入四氢呋喃(10 ml)中,氩气保护下降温至-60 o
c,滴加仲丁基锂的正己烷溶液(1.3 m,4.5 ml,5.88 mmol),-50 o
c搅拌1小时,加入n, n-二甲基甲酰胺(0.32 g,4.41 mmol),-50 o
c搅拌1小时,室温搅拌1小时。反应液用饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10/1)得到标题化合物(340 mg,41 %)。ms: m/z [m+h-boc]
+ =184。
[0192] 8-(羟甲基)-3-氧杂-9-氮杂螺[5. 5]十一烷-9-羧酸叔丁酯
室温下,将8-甲酰基-3-氧杂-9-氮杂螺[5. 5]十一烷-9-羧酸叔丁酯(340 mg,1.2 mmol)加入四氢呋喃(10 ml)中,搅拌下加入硼氢化钠(40 mg,1.2 mmol),室温搅拌0.5小时。反应液用饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩得到标题化合物(340 mg,99 %)。ms: m/z [m+h-boc]
+ =186。
[0193]
(9-(6-氯-2-甲氧基嘧啶-4-基)-3-氧杂-9-氮杂螺[5. 5]十一烷-8-基)甲醇室温下,将8-(羟甲基)-3-氧杂-9-氮杂螺[5. 5]十一烷-9-羧酸叔丁酯(340 mg,1.2 mmol)加入三氟乙酸(1 ml)和二氯甲烷(4 ml)中,室温搅拌5分钟。用碳酸钠固体将反应体系ph调节至8-9。反应液浓缩,向残留物中依次加入4, 6-二氯-2-甲氧基嘧啶(0.32 g,1.78 mmol)、碳酸钠(0.63 g,5.95 mmol)和乙腈(30 ml)中,85 o
c搅拌过夜。反应液过滤,滤液浓缩,柱层析纯化(二氯甲烷/甲醇 = 20 /1)得到标题化合物(210 mg,54 %)。ms: m/z [m+h]
+ =328。
[0194] 3'-氯-2, 3, 5, 6, 6', 7', 9a', 10'-八氢-1'h, 9'h-螺[吡喃-4, 8'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮室温下,将(9-(6-氯-2-甲氧基嘧啶-4-基)-3-氧杂-9-氮杂螺[5. 5]十一烷-8-基)甲醇(141 mg,0.43 mmol)和三乙胺(0.13 g,1.29 mmol)加入二氯甲烷(12 ml)中,搅拌下加入甲磺酰酐(74 mg,0.65 mmol),室温搅拌20分钟。用碳酸钠固体将反应液ph调节至8-9。反应液浓缩,向残留物中依次加入水(8 ml)和碳酸钾(0.29 g,2.1 mmol)。室温搅拌30分钟。二氯甲烷提取,有机相浓缩,制备薄层色谱分离(二氯甲烷/甲醇 = 20/1)得到标题化合物(60 mg,47 %)。ms: m/z [m+h]
+ =296。
[0195]
中间体151-(叔丁基)4-乙基 4-甲基哌啶-1, 4, 4-三羧酸室温下,将1-(叔丁基)4-乙基哌啶-1, 4-二羧酸酯(45.0 g,175.1 mmol )加入无
水四氢呋喃(180 ml)中,氩气保护下降温至-60 o
c,搅拌下加入二异丙基氨基锂的四氢呋喃溶液/正庚烷溶液(2 m,131 ml,262 mmol),-60 o
c搅拌0.5小时,加入氯甲酸甲酯(18.2 g,192.6 mmol),-60 o
c搅拌30分钟,室温搅拌两小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 5/1)得到标题化合物(41 g,91 %)。ms: m/z [m+h-boc]
+ =216。
[0196] 4, 4-双(羟甲基)哌啶-1-羧酸叔丁酯室温下,将1-(叔丁基)4-乙基 4-甲基哌啶-1, 4, 4-三羧酸(10.0 g,31.7 mmol )加入四氢呋喃(50 ml)和甲醇(50 ml)中,反应升温至50 o
c,控温50 o
c-60 o
c分批次加入硼氢化钠(6.0 g,158.5 mmol),60 o
c搅拌1小时。反应液用饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩得到标题化合物粗品(8 g,103 %)。ms: m/z [m+h-boc]
+ =146。
[0197] 4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)哌啶-1-羧酸叔丁酯室温下,将4, 4-双(羟甲基)哌啶-1-羧酸叔丁酯(45.0 g,183 mmol)和咪唑(37.9 g,549 mmol)依次加入二氯甲烷(100 ml)中,搅拌下加入叔丁基二甲基氯硅烷(55.2 g,366 mmol),室温搅拌2小时。反应液倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10/1)得到标题化合物(66 g,77 %)。ms: m/z [m+h-boc]
+ =373。
[0198]
4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-2-甲酰基哌啶-1-羧酸叔丁酯室温下,将 4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)哌啶-1-羧酸叔丁酯(20.0 g,42.28 mmol)和四甲基乙二胺(7.35 g,64.42 mmol)加入无水四氢呋喃(120 ml)中,氩气保护下降温至-60 o
c,控温-50 o
c以下滴加仲丁基锂的环己烷溶液(1.3 m,71.5 ml,93.0 mmol),-50 o
c搅拌1小时,加入n, n-二甲基甲酰胺(4.56 g,63.42 mmol),-50 o
c搅拌1 h,室温搅拌2小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10 /1)得到标题化合物(12 g,57 %)。ms: m/z [m+h-boc]
+ =402。
[0199] (4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)哌啶-2-基)甲醇
室温下,将4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-2-甲酰基哌啶-1-羧酸叔丁酯(4.0 g,8.0 mmol)加入三氟乙酸的二氯甲烷溶液(10%,40 ml)中,室温搅拌5分钟。反应液用碳酸钾固体调节至8-9,过滤,滤液浓缩。将残留物加入甲醇(20 ml)中,搅拌下加入硼氢化钠(907 mg,24.0 mmol),室温搅拌1小时。反应液用饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩得到标题化合物粗品(4 g,124 %)。ms: m/z [m+h]
+ =404。
[0200]
(4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-1-(6-氯-2-甲氧基嘧啶-4-基)哌啶-2-基)甲醇室温下,将(4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)哌啶-2-基)甲醇的粗品(3.0 g,7.44 mmol)、4, 6-二氯-2-甲氧基嘧啶(1.32 g,7.44 mmol)和碳酸钠固体(1.58 g,14.88 mmol)加入乙腈(30 ml)中,90 o
c搅拌过夜。过滤,滤液浓缩,柱层析纯化(石油醚/乙酸乙酯 = 10/1~5/1)得到标题化合物(1 g,25 %)。ms: m/z [m+h]
+ =546。
[0201] 8, 8-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-氯-6, 7, 8, 9, 9a, 10-六氢-1h-吡啶并[1', 2': 3, 4]咪唑[1, 2-c]嘧啶-1-酮室温下,将(4, 4-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-1-(6-氯-2-甲氧基嘧啶-4-基)哌啶-2-基)甲醇(1.0 g,2.0 mmol)加入二氯甲烷(20 ml)中,搅拌下将三乙胺(303 mg,3.0 mmol)和甲基磺酰氯(273 mg,2.4 mmol)依次加入反应液,室温搅拌20分钟。反应液浓缩,向残留物中加入水(16 ml),搅拌下用碳酸钾固体将体系ph调节至9-10,室温搅拌30分钟。二氯甲烷提取,有机相浓缩,柱层析纯化(二氯甲烷/甲醇 = 20 /1)得到标题化合物(450 mg,48 %)。ms: m/z [m+h]
+ =514。
[0202] 8, 8-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6, 7, 8, 9, 9a, 10-六氢-1h-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶-1-酮
室温下,依次将8, 8-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-氯-6, 7, 8, 9, 9a, 10-六氢-1h-吡啶并[1', 2': 3, 4]咪唑[1, 2-c]嘧啶-1-酮(450 mg,0.88 mmol)、(3-氟-4-[(2-(三氟甲基)吡啶-4-基)氧基]苯基)甲醇(0.28 g,0.97 mmol)和碳酸铯(0.86 g,2.64 mmol)加入甲苯(20 ml)中,110 o
c搅拌过夜。反应液浓缩后倒入水中,二氯甲烷提取,有机相浓缩,柱层析纯化(二氯甲烷/甲醇 = 20/1)得到标题化合物(670 mg,99 %)。ms: m/z [m+h]
+ =765。
[0203] 3-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-8, 8-双(羟甲基)-6, 7, 8, 9, 9a, 10
ꢀ‑
六氢-1h-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶-1-酮室温下,将8, 8-双(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6, 7, 8, 9, 9a, 10-六氢-1h-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶-1-酮(670 mg,0.88 mmol)加入无水四氢呋喃(10 ml)中,搅拌下加入四丁基氟化铵的四氢呋喃溶液(1 m,2.2 ml,2.2 mmol),室温搅拌1 小时。反应液倒入饱和氯化铵水溶液中,乙酸乙酯提取,有机相浓缩,柱层析纯化(二氯甲烷/甲醇 = 20/1~10/1)得到标题化合物(350 mg,74 %)。ms: m/z [m+h]
+ =537。
[0204]
(3-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-8-(羟甲基)-1-氧代-6, 7, 8, 9, 9a, 10-六氢-1h-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶-8-基)甲磺酸甲酯室温下,将3-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-8, 8-双(羟甲基)-6, 7, 8, 9, 9a, 10
ꢀ‑
六氢-1h-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶-1-酮(100 mg,0.19 mmol)加入二氯甲烷(10 ml)中,氩气保护下降温至0 o
c,搅拌下依次加入三乙胺(23.07 mg,0.23 mmol)和甲基磺酰氯(13.06 mg,0.11 mmol )的二氯甲烷(1 ml)溶液,室温搅拌15分钟。反应液倒入水中,二氯甲烷提取,有机相浓缩,制备薄层色谱分离(二氯甲烷/甲醇 = 10/1)得到标题化合物(60 mg,51 %)。ms: m/z [m+h]
+ =615。
[0205]
中间体165-甲酰基-2-(3-(三氟甲基)苯氧基)苄腈
室温下,将2-氟-5-甲酰基苄腈(15.0 g,0.1 mol)、3-(三氟甲基)苯酚(16.0 g,0.1 mol)和碳酸钾(13.8 g,0.1 mol)加到dmf(100 ml)中,105 o
c搅拌8小时,反应液倒入水中,用二氯甲烷萃取,有机相浓缩,向残留物中加入乙醇(50 ml),室温搅拌30分钟,过滤,滤饼浓缩干得到标题化合物(22.1 g,76%)。1h nmr(400 mhz, dmso-d6)d 9.96(s, 1 h), 8.49(s, 1 h), 8.14(br.s, 1 h), 7.88(d, 2 h), 7.47(d, 2 h), 7.19(d, 1 h)。
[0206] 5-(羟甲基)-2-(3-(三氟甲基)苯氧基)苄腈室温下,将5-甲酰基-2-(3-(三氟甲基)苯氧基)苄腈(24.3 g,83.44 mmol)加入甲醇(250 ml)中,分批加入硼氢化钠(4.86 g,127.9 mmol),室温反应0.5小时。反应液倒入水中,用二氯甲烷萃取,有机相浓缩得到标题化合物(24.2 g,99 %)。1h nmr(400 mhz, dmso-d6)d 7.80(s, 1 h), 7.73
ꢀ‑ꢀ
7.53(m, 3 h), 7.52
ꢀ‑ꢀ
7.27(m, 2 h), 7.09(br.s, 1 h), 5.41(s, 1 h), 4.51(s, 2 h)。
[0207]
参考下表,采用“原料”一栏所述化合物为原料,参照中间体16的制备方法制备以下中间体17~23。
[0208]
[m+h]
+
=208。
[0216] 2-(二氟甲基)-4-甲氧基吡啶将4-溴-2-(二氟甲基)吡啶(11.18g,53.76mmol)、甲醇钠(5.81g,107.52mmol)加入甲醇(100ml)中。90oc搅拌过夜。反应液降温至室温并倒入水(300ml)中,乙酸乙酯提取。有机相浓缩得到标题化合物粗品直接用于下一步反应。ms:m/z[m+h]
+
=160。
[0217] 2-(二氟甲基)吡啶-4-醇将2-(二氟甲基)-4-甲氧基吡啶(9.54g,59.95mmol)加入氢溴酸(40%水溶液,58ml)中。90oc搅拌2天。反应液浓缩,将残留物加水稀释,搅拌下加入碳酸氢钠固体直到体系没有气泡产生为止,乙酸乙酯提取,有机相浓缩,柱层析纯化(石油醚/乙酸乙酯=10/1~1/1)得到标题化合物(2.5g,三步总收率26%)。ms:m/z[m+h]
+
=146。
[0218]
4-((2-(二氟甲基)吡啶-4-基)氧基)-3-氟苯甲醛将3,4-二氟苯甲醛(350mg,2.46mmol)、2-(二氟甲基)吡啶-4-醇(357mg,2.46mmol)和碳酸钾固体(1019mg,7.38mmol)加入到n,n-二甲基甲酰胺(5ml)中,120oc搅拌3小时。反应液倒入水中,乙酸乙酯提取,有机相浓缩,制备薄层色谱分离得到标题化合物(510mg,78%)。ms:m/z[m+h]
+
=268。
[0219] (4-((2-(二氟甲基)吡啶-4-基)氧基)-3-氟苯基)甲醇将4-((2-(二氟甲基)吡啶-4-基)氧基)-3-氟苯甲醛(505mg,1.89mmol)加入无水乙醇(5ml)中,搅拌下加入nabh
4(
140mg,3.78mmol),室温搅拌2小时。反应液倒入水中,二氯甲烷提取,有机相浓缩得到标题化合物(381mg,75%)。ms:m/z[m+h]
+
=271。
实施例
[0220]
实施例1方法a3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6',7',9a',10'-四氢-1'h,9'h-螺[环丙烷-1,8'-吡啶并[1',2':3,4]咪唑并[1,2-c]嘧啶]-1'-酮
室温下,将3'-氯-6', 7', 9a', 10'-四氢-1'h, 9'h-螺[环丙烷-1, 8'-吡啶基[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮(30 mg,0.12 mmol)、(3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苯基)甲醇(50 mg,0.17 mmol)和氢化钠(7 mg,0.18 mmol)加入乙腈(5 ml)中,室温搅拌1小时。反应液饱和氯化铵水溶液淬灭后倒入水中,乙酸乙酯提取,有机相浓缩,制备薄层色谱纯化得到标题化合物(9 mg,15%)。1h nmr(400 mhz, cdcl3)d 8.56(d, j = 5.4 hz, 1 h), 7.32(d, j = 11.2 hz, 1 h), 7.28
ꢀ‑ꢀ
7.14(m, 3 h), 6.95(d, j = 3.4 hz, 1 h), 5.43(s, 2 h), 5.04(s, 1 h), 4.35
ꢀ‑ꢀ
4.13(m, 1 h), 4.01
ꢀ‑ꢀ
3.84(m, 1 h), 3.67(dd, j = 7.3, 11.2 hz, 1 h), 3.53(dd, j = 3.9, 12.7 hz, 1 h), 3.22
ꢀ‑ꢀ
3.07(m, 1 h), 2.09
ꢀ‑ꢀ
1.92(m, 2 h), 1.11(d, j = 12.7 hz, 1 h), 0.88(d, j = 12.7 hz, 1 h), 0.53
ꢀ‑ꢀ
0.38(m, 4 h); ms: m/z [m+h]
+ =503。
[0221] 实施例 2方法b3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-8', 9', 9a', 10'-四氢-1'h, 7'h-螺[环丙烷-1, 6'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将3'-氯-8', 9', 9a', 10'-四氢-1'h, 7'h-螺[环丙烷-1, 6'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮(80 mg,0.3 mmol)、(3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苯基)甲醇(86 mg,0.3 mmol)和碳酸铯固体(195 mg,0.6 mmol)加入甲苯(10 ml)中,100 o
c搅拌过夜。反应液过滤,滤液浓缩,制备薄层色谱分离得到标题化合物(50 mg,33%)。1h nmr(400 mhz, cdcl3)d 8.55(d, j = 5.4 hz, 1 h), 7.34(d, j = 10.8 hz, 1 h), 7.27
ꢀ‑ꢀ
7.25(m, 1 h), 7.23
ꢀ‑ꢀ
7.12(m, 2 h), 6.95(d, j = 5.4 hz, 1 h), 5.41(s, 2 h), 5.35(s, 1 h), 3.94
ꢀ‑ꢀ
3.84(m, 3 h), 2.04(t, j = 12.5 hz, 1 h), 1.85(br. s., 1 h), 1.79
ꢀ‑ꢀ
1.71(m, 1 h), 1.53(br. s., 1 h), 1.41
ꢀ‑ꢀ
1.29(m, 1 h), 1.20
ꢀ‑ꢀ
1.13(m, 1 h), 0.96
ꢀ‑ꢀ
0.84(m, 2 h), 0.78
ꢀ‑ꢀ
0.68(m, 1 h), 0.58
ꢀ‑ꢀ
0.48(m, 1 h); ms: m/z [m+h]
+ =503。
[0222]
实施例 3方法c3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6', 7', 9a', 10'-四氢-1'h, 9 'h-螺[氧杂环丁烷-3, 8'-吡啶并[1', 2': 3, 4]咪唑并[1, 2-c]嘧啶]-1'-酮
9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1'-酮室温下,将3'-((3-氟-4-((2-(三氟甲基)吡啶-4-基)氧基)苄基)氧基)-6', 7', 9a', 10'-四氢-1'h, 9'h-螺环[环丁烷-1, 8'-吡啶[1', 2': 3, 4]咪唑[1, 2-c]嘧啶]-1', 3-二酮(33 mg,0.12 mmol)和甲胺盐酸盐(13 mg,0.2 mmol)加入甲醇(3 ml)中,搅拌下加入氰基硼氢化钠(21 mg,0.33 mmol),室温搅拌过夜。反应液用水淬灭,二氯甲烷萃取,有机相浓缩,制备薄层色谱纯化得到标题化合物(9 mg,25%)。1h nmr(400 mhz, cdcl3)d 8.58 (d, j = 5.4 hz, 1 h), 7.33 (d, j = 10.8 hz, 1 h), 7.27
ꢀ‑ꢀ
7.25 (m, 1 h), 7.23
ꢀ‑ꢀ
7.15 (m, 2 h), 6.97 (br. s., 1 h), 5.42 (br. s., 2 h), 5.05 (s, 1 h), 4.29
ꢀ‑ꢀ
4.19 (m, 1 h), 3.83 (br. s., 1 h), 3.76
ꢀ‑ꢀ
3.61 (m, 2 h), 3.51 (d, j = 12.2 hz, 1 h), 3.16
ꢀ‑ꢀ
2.97 (m, 1 h), 2.58 (br. s., 3 h), 2.52
ꢀ‑ꢀ
2.34 (m, 3 h), 2.30
ꢀ‑ꢀ
2.16 (m, 2 h), 2.12
ꢀ‑ꢀ
1.97 (m, 2 h), 1.67
ꢀ‑ꢀ
1.59 (m, 1 h); ms: m/z [m+h]
+ =546。
[0225]
下表所列的实施例6-34通过类似于实施例1-5所述的步骤制备,起始于相应的中间体:
[0226]
生物测试和数据本发明的化合物为lp-pla2抑制剂,可用于治疗和预防lp-pla2介导的疾病。本发明的化合物的生物活性可使用用于确定化合物作为lp-pla2抑制剂的活性的任何合适的测试,以及组织和体内模型进行测定。
[0227]
各化合物的生物活性数据以至少一个实验或多个实验的平均值报告。应理解本发明描述的数据可根据实施实验的人所使用的具体条件和方法而存在合理的变化。
[0228] 脂蛋白-相关磷脂酶a2 (lp-pla2)人血浆测定。
[0229]
人血浆测定利用paf(磷脂酰胆碱)的硫酯类似物,其中水解导致形成包含游离巯基的磷脂。通过与cpm(7-二乙基氨基-3-(4
’‑
马来酰亚胺基苯基)-4-甲基香豆素)反应来连续测定巯基的量,该cpm为一种在巯基的michael加成后荧光增加的马来酰亚胺。该测定可以检测在人血浆的lp-pla2的活性,如通过lp-pla2抑制剂的特异性抑制所确定。
[0230]
thio-paf测定作为淬灭的75μl测定进行。通过在96孔微量板上在纯dmso中制备所述各个化合物的1:3(体积)系列稀释液来准备化合物源板。通过rainin多道移液器将化合物源板上的3μl化合物转移入预先添加57μl测定缓冲液的 96孔微量板中,将源板上的化合物进行20倍稀释。上述测试缓冲液含有由50mm hepes,ph 7.4、150mm nacl、1mm chaps组成。通过rainin多道移液器将进行20倍稀释后的化合物转移1μl入预先添加等分和解冻的40μl混合人血浆的96孔greiner 655076(黑色)微量板中。板在酶标板振荡器振荡20秒混匀。室温下30分钟的预孵育后,通过rainin多道移液器将10μl的底物溶液添加到96孔greiner 655076(黑色)微量板中,所述底物溶液含有由50mm hepes,ph 7.4、150mmnacl、1mm chaps组成的测定缓冲液中的2.5mm 2-thio-paf[来自乙醇母液]、32 μm cpm[来自dmso母液]和3.2mm nem(n-乙基马来酰亚胺)[每次实验在dmso中新制备]。2分钟后,用25μl的5%三氟乙酸(tfa)水溶液淬灭反应。板以2000rpm离心1分钟。使用biotek synergy h1(h1mf)酶标仪以ex:380/em:485读板。使用graphpad prism 6.0和excel进行ic
50
数据、曲线和qc分析。
[0231]
实施例测定活性
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1