一种羰基二咪唑合成终点的控制方法与流程
1.本发明属于有机化合物合成技术领域,尤其涉及一种高品质羰基二咪唑合成终点的控制方法。
背景技术:
2.羰基二咪唑是一种具有较强反应活性的化合物,它可与—cooh、—nh2、—oh等官能团进行反应合成许多用一般方法难以得到的酮、酯、脲等化合物。由于在反应过程中能够保持分子构型不变,不形成氢卤酸,因而在生化合成中具有非常重要的意义。羰基二咪唑作为强活化剂,是重要的医药中间体,广泛用作酶和蛋白质粘合剂、抗生素类合成,特别是作为合成多肽化合物的键合剂。随着生物化学领域的飞速发展,n,n
′‑
羰基二咪唑已备受市场青睐,国内外市场需求量日益增大。
3.羰基二咪唑的合成方法研究主要分为以下4种:
4.1、以甲苯、邻二甲苯,四氢呋喃等为溶剂,光气和咪唑反应生成羰基二咪唑。
5.咪唑同时作为原料和缚酸剂,反应结束时,至少有一半的咪唑为盐酸盐存在,单批次咪唑的利用率不高。咪唑盐酸盐用无机强碱中和,经洗涤、干燥后,回收使用。
6.2、在咪唑-光气法的基础上,引入碱性大于咪唑的有机碱。
7.一般以三正丁胺等有机碱为反应缚酸剂,充分利用了体系中的咪唑原料,提高了单批次咪唑的利用率。反应结束,过滤出产品后,含有机碱盐酸盐的溶剂,用无机强碱中和,经除水后,可投入下一批次使用。但是,反应体系中需加入大量有机碱,以杂质的形式存在于最终产品中。在通光的过程中,有机碱易变色,会影响最终产品的颜色。
8.3、以三甲基硅咪唑为中间体
9.咪唑先与六甲基二硅胺烷或三甲基甲硅烷基氯等反应得到三甲基硅咪唑,中间产物再与光气反应得到产物。但是,该方法路线长,产品杂质含量高。
10.4、以咪唑和芳族碳酸酯为原料,在一定的温度下反应合成羰基二咪唑。但是,该方法收率、含量均不高,产品提纯困难。
11.申请人在研究中发现,在光气加入量合适的条件下,方法1合成的羰基二咪唑产品品质相对最高,仅光气加入量对产品的品质影响比较大,光气不足时产品中咪唑含量高,光气过量时产品易吸潮溶解。因此,研究合成终点的控制方法是合成高品质羰基二咪唑的关键。
技术实现要素:
12.本发明要解决的技术问题是克服现有技术的不足,提供一种高品质羰基二咪唑合成终点控制方法。
13.为解决上述技术问题,本发明采用以下技术方案。
14.一种羰基二咪唑合成终点的控制方法,包括以下步骤:在n2保护下,将原料咪唑、溶剂四氢呋喃混合,在搅拌下加入过量光气进行反应,控制反应温度为5℃~10℃,光气加
入完成后,继续搅拌下保温30min~40min,再静置10min~15min,取反应液上层清液定量测定氯离子质量百分含量,根据氯离子质量百分含量,在搅拌下定量回加咪唑,加完后,再次测定反应液清液中的氯离子,无氯离子即为反应到达终点,反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂,得到羰基二咪唑产品。
15.上述的羰基二咪唑合成终点的控制方法,优选的,所述原料咪唑与光气的摩尔比为4∶1.2~1.3。
16.上述的羰基二咪唑合成终点的控制方法,优选的,再次测定反应液清液中的氯离子时,若存在氯离子,则取反应液清液定量测定氯离子质量百分含量,根据氯离子质量百分含量,在搅拌下定量回加咪唑,加完后,继续测定反应液清液中的氯离子,如此反复,直至无氯离子即为反应到达终点。
17.上述的羰基二咪唑合成终点的控制方法,优选的,所述回加咪唑时,以咪唑的四氢呋喃溶液的形式进行回加,咪唑的四氢呋喃溶液中咪唑的质量分数为10%。
18.上述的羰基二咪唑合成终点的控制方法,优选的,所述光气以光气的四氢呋喃溶液的形式加入。
19.上述的羰基二咪唑合成终点的控制方法,优选的,回加咪唑的四氢呋喃溶液的量按下式进行计算:
[0020][0021]
其中,x为回加咪唑的四氢呋喃溶液的量,单位为g,cl%为检测出的氯离子质量百分含量,m为测定氯离子质量百分含量前加入的所有的咪唑、光气、四氢呋喃的质量之和,单位为g,即第一次回加咪唑的四氢呋喃溶液时,m为测定氯离子质量百分含量前加入的原料咪唑、溶剂四氢呋喃、光气的四氢呋喃溶液的质量之和,第二次及第二次以后回加咪唑的四氢呋喃溶液时,m为测定氯离子质量百分含量前加入的原料咪唑、溶剂四氢呋喃、光气的四氢呋喃溶液、咪唑的四氢呋喃溶液的质量之和。
[0022]
上述的羰基二咪唑合成终点的控制方法,优选的,所得羰基二咪唑产品的收率为90.0%~92.3%,含量为99.0%~99.6%。
[0023]
本发明中,对四氢呋喃的量不进行限定,能基本溶解即可,可根据实际情况确定。
[0024]
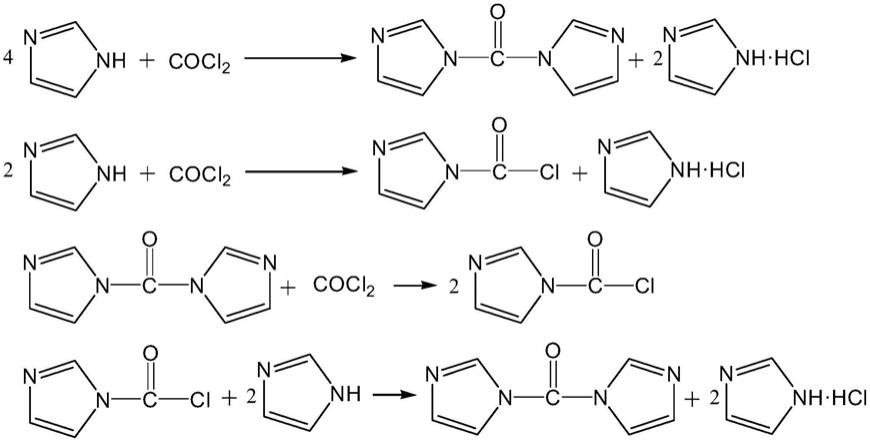
本发明中,反应过程中的相关反应式如下所示:
[0025][0026]
本发明的原理在于:
[0027]
1、咪唑盐酸盐不溶解于四氢呋喃,且颗粒较大,稍加静置即可得到沉淀及澄清溶液。在光气不足量的条件下取清液,检测不到氯离子。
[0028]
2、光气过量后,反应产生酰氯并溶解于四氢呋喃。取反应液清液可测出氯离子含量。
[0029]
3、回加咪唑时,酰氯与一半的咪唑继续反应生成产品,一半的咪唑生成盐酸盐,终点时反应清液中无氯离子。
[0030]
4、回加咪唑的四氢呋喃溶液的量x按式(1)计算
[0031][0032]
式(1)中:
[0033]
cl%——检测出的氯离子质量百分含量;
[0034]
m——加入的所有的咪唑、四氢呋喃、光气的质量之和,单位为g;
[0035]
2——咪唑相对氯离子反应的摩尔比;
[0036]
68.08——咪唑的摩尔质量,单位为g/mol;
[0037]
35.5——氯元素的摩尔质量,单位为g/mol;
[0038]
0.1——咪唑的质量换算成10wt%咪唑的四氢呋喃溶液质量。
[0039]
与现有技术相比,本发明的优点在于:
[0040]
(1)本发明的方法可以准确地判定反应终点,以提高产品含量。
[0041]
(2)本发明的方法无需加入可能影响产品品质的其他原料。
[0042]
(3)本发明的方法产品收率高,收率达90.0%~92.3%。
[0043]
(4)本发明的方法所得产品含量高,含量达99.0%~99.6%。
[0044]
(5)本发明的方法使用的其他原料和溶剂均便宜易得,工艺简单,有利于工业化生产。
具体实施方式
[0045]
以下结合具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的
保护范围。以下实施例中所采用的材料和仪器均为市售,氯离子含量均指氯离子的质量百分含量。
[0046]
实施例1:
[0047]
一种本发明的羰基二咪唑合成终点的控制方法,包括以下步骤:
[0048]
在n2保护下,将41.3g(0.6mol,99%)咪唑和400g四氢呋喃投入到带冷凝管的1000ml四口瓶中,开启搅拌。控制反应温度10℃,1小时内滴入含光气的四氢呋喃溶液118g,含光气的四氢呋喃溶液中含有18.0g(0.1818mol)光气。光气加入完成,继续搅拌下保温30min后,关闭搅拌静置10min,取反应液上层清液定量测定氯离子含量为0.018%。根据氯离子含量,搅拌中定量滴加质量分数为10%的咪唑的四氢呋喃溶液3.86g。滴加完成后,再次测定反应液清液中的氯离子,已无氯离子,则反应到达终点。反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂,得到羰基二咪唑产品22.62g,含量99.51%,收率为91.48%。
[0049]
实施例2:
[0050]
一种本发明的羰基二咪唑合成终点的控制方法,包括以下步骤:
[0051]
在n2保护下,将41.3g(0.6mol,99%)咪唑和400g四氢呋喃投入到带冷凝管的1000ml四口瓶中,开启搅拌。控制反应温度10℃,1小时内滴入含有19.3g(0.1949mol)光气的光气的四氢呋喃溶液119.4g。光气加入完成,保温30min后,关闭搅拌静置10min,取反应液上层清液定量测定氯离子含量为0.031%。根据氯离子含量,搅拌中定量加入10%咪唑的四氢呋喃溶液6.67g。滴加完成后,再次测定反应液清液中氯离子,无氯离子,反应到达终点。反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂,得到羰基二咪唑产品22.50g,含量99.14%,收率为90.04%。
[0052]
实施例3:
[0053]
一种本发明的羰基二咪唑合成终点的控制方法,包括以下步骤:
[0054]
在n2保护下,将41.3g(0.6mol,99%)咪唑和400g四氢呋喃投入到带冷凝管的1000ml四口瓶中,开启搅拌。控制反应温度10℃,1小时内滴入含有19.1g(0.1929mol)光气的光气——四氢呋喃溶液98g。光气加入完成,保温30min后,关闭搅拌静置10min,取反应液上层清液定量测定氯离子含量为0.034%。根据氯离子含量,搅拌中定量加入10wt%咪唑的四氢呋喃溶液7.03g。滴加完成后,再次测定反应液清液中氯离子,无氯离子,反应到达终点。反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂,得到羰基二咪唑产品23.09g,含量99.02%,收率为92.21%。
[0055]
实施例4:
[0056]
一种本发明的羰基二咪唑合成终点的控制方法,包括以下步骤:
[0057]
在n2保护下,将41.3g(0.6mol,99%)咪唑和500g四氢呋喃投入到带冷凝管的1000ml四口瓶中,开启搅拌。控制反应温度10℃,1小时内滴入含有18.2g(0.1838mol)光气的光气——四氢呋喃溶液126g。光气加入完成,保温30min后,关闭搅拌静置10min,取反应液上层清液定量测定氯离子含量为0.015%。根据氯离子含量,搅拌中定量加入10%咪唑的四氢呋喃溶液3.84g。滴加完成后,再次测定反应液清液中氯离子,无氯离子,反应到达终点。反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂得到羰基二咪唑产品22.65g,含量99.41%,收率为91.52%。
[0058]
实施例5:
[0059]
一种本发明的羰基二咪唑合成终点的控制方法,包括以下步骤:
[0060]
在n2保护下,将41.3g(0.6mol,99%)咪唑和500g四氢呋喃投入到带冷凝管的1000ml四口瓶中,开启搅拌。控制反应温度10℃,1小时内滴入含有18.6g(0.1879mol)光气的光气——四氢呋喃溶液130g。光气加入完成,保温30min后,关闭搅拌静置10min,取反应液上层清液定量测定氯离子含量为0.022%。根据氯离子含量,搅拌中定量加入10%咪唑的四氢呋喃溶液5.66g。滴加完成后,再次测定反应液清液中氯离子,无氯离子,反应到达终点。反应结束后,在n2保护下,过滤出咪唑盐酸盐,滤液在室温下蒸除溶剂得到羰基二咪唑产品22.52g,含量99.22%,收率为90.42%。
[0061]
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制。虽然本发明已以较佳实施例揭示如上,然而并非用以限定本发明。任何熟悉本领域的技术人员,在不脱离本发明的精神实质和技术方案的情况下,都可利用上述揭示的方法和技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同替换、等效变化及修饰,均仍属于本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1