3-氯-β-亚甲基苯乙醇类化合物及其中间体各自的制备方法与流程
1.本发明涉及有机合成技术领域,具体而言,涉及一种3-氯-β-亚甲基苯乙醇类化合物及其中间体各自的制备方法。
背景技术:
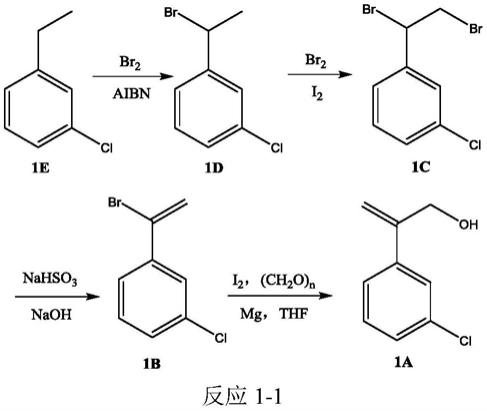
2.3-氯-β-亚甲基苯乙醇作为制备除草剂茚草酮的主要原料而备受关注,由于其结构简单,相对合成路线繁多,目前主要合成路线包含如下两种方案。一种为日本三菱公司公开的合成方法,是以间氯乙苯(1e)为原料,经与溴素二溴代得到(1c),再脱一分子溴化氢得到(1b),最后1b制备格氏试剂与多聚甲醛亲核加成反应并水解得到(1a),如反应1-1所示:
[0003][0004]
以上方法的缺陷是步骤长、条件要求苛刻、纯化难度大以及得到大量副产物溴化氢,环境污染较重,成本较高,不利于工业化大规模生产。
[0005]
制备3-氯-β-亚甲基苯乙醇的另一种方法是,间氯卤苯(2b)制备格氏与丙炔醇在碘化亚铜催化下反应,得到3-氯-β-亚甲基苯乙醇(1a),如反应1-2所示:
[0006][0007]
但由于间氯卤苯制备格氏时产生双格氏化反应而引入副产物,导致纯化难度大、收率低,同时格氏试剂自偶联反应形成二氯联苯,多氯联苯会造成环境污染;且丙炔醇为剧毒化合物,运输、储存都比较受限,以上两种原因导致该方法很少使用。
技术实现要素:
[0008]
本发明的主要目的在于提供一种3-氯-β-亚甲基苯乙醇类化合物及其中间体各自
的制备方法,以解决现有技术中除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇的制备方法复杂、成本较高的问题。
[0009]
为了实现上述目的,根据本发明的一个方面,提供了一种3-氯-β-亚甲基苯乙醇类化合物的中间体的制备方法,该制备方法包括:步骤s1,将3-氯-苯甲酰氯类化合物与氯甲烷格氏试剂进行亲核加成反应后水解,得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系;步骤s2,对中间体体系的3-氯-α,α-二甲基苯甲醇类化合物进行脱水反应,得到3-氯-β-亚甲基苯乙醇类化合物的中间体,3-氯-β-亚甲基苯乙醇类化合物的中间体为1-氯-3-(1-甲基乙烯基)苯类化合物,其中,3-氯-苯甲酰氯类化合物、3-氯-α,α-二甲基苯甲醇类化合物、1-氯-3-(1-甲基乙烯基)苯类化合物的结构式依次如下:
[0010][0011]
r选自h、c1~c
10
的烷基、c1~c
10
的烷氧基、f、cl、三氟甲基中的任意一种。
[0012]
进一步地,上述r选自h、c1~c6的烷基、c1~c6的烷氧基中的任意一种,优选r选自h、甲基、乙基、正丙基、甲氧基、乙氧基中的任意一种,进一步地,优选r为h。
[0013]
进一步地,上述3-氯-苯甲酰氯类化合物与氯甲烷格氏试剂的摩尔比为1:2.2~3.0。
[0014]
进一步地,上述亲核加成反应的温度为20~35℃,优选亲核加成反应的时间为2~3小时。
[0015]
进一步地,上述水解在10~15wt%的盐酸溶液中进行,优选水解的温度为0~30℃。
[0016]
进一步地,上述亲核加成反应的溶剂选自四氢呋喃、2-甲基四氢呋喃、乙醚、甲苯中的任意一种或多种。
[0017]
进一步地,上述脱水反应在脱水剂作用下进行,脱水剂为3-氯-α,α-二甲基苯甲醇类化合物的1~5wt%,优选脱水剂选自甲苯-4-磺酸、蒙脱石、硫酸高铈、硫酸锌中的任意一种或多种。
[0018]
进一步地,上述脱水反应中还包括有抗氧化剂,抗氧化剂为3-氯-α,α-二甲基苯甲醇类化合物的0.1~0.5wt%,优选抗氧化剂选自2,6-二叔丁基-4-甲基苯酚、对叔丁基邻苯二酚、对苯二酚中的任意一种或多种。
[0019]
进一步地,上述脱水反应的溶剂选自甲苯、二甲苯、环己烷、甲基环己烷、正庚烷、石油醚中的任意一种或多种,优选脱水反应的温度为80~140℃,优选脱水反应的时间为8~10h。
[0020]
根据本发明的另一方面,提供了一种3-氯-β-亚甲基苯乙醇类化合物的制备方法,该制备方法包括:通过前述的制备方法得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物,将1-氯-3-(1-甲基乙烯基)苯类化合物进行催化氧化反
应,得到3-氯-β-亚甲基苯乙醇类化合物,3-氯-β-亚甲基苯乙醇类化合物的结构式为:
[0021][0022]
其中,r选自h、c1~c
10
的烷基、c1~c
10
的烷氧基、f、cl、三氟甲基中的任意一种或多种。
[0023]
进一步地,上述催化氧化反应在氧化剂、氧化催化剂及醇类溶剂中进行,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯类化合物的质量比为0.1~0.5:1;氧化催化剂包括过渡金属氧化物与γ-al2o3载体,过渡金属氧化物包括氧化锰、氧化钴和氧化铈,氧化锰、氧化钴、氧化铈与γ-al2o3载体的质量比为0.001~0.05:0.001~0.05:0.001~0.01:1;氧化剂与1-氯-3-(1-甲基乙烯基)苯类化合物的摩尔比为1.2~1.5:1,优选氧化剂选自含氧气体、双氧水、次氯酸钠中的任意一种或多种,含氧气体中的氧气含量为1~50%;优选醇类溶剂选自甲醇、乙醇、异丙醇中的任意一种或多种;优选催化氧化反应的温度为20~35℃,优选催化氧化反应的时间为8~10h。
[0024]
应用本发明的技术方案,本技术采用廉价易得的3-氯-苯甲酰氯类化合物为原料,先与氯甲烷格氏试剂进行亲核加成反应并水解得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系。3-氯-α,α-二甲基苯甲醇类化合物经脱水反应得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物。该方法步骤少、反应条件温和、成本低。并且将该制备方法得到的1-氯-3-(1-甲基乙烯基)苯类化合物进行催化氧化即可得到目标产物3-氯-β-亚甲基苯乙醇类化合物,可见,本技术仅需要经过三步反应,就可以得到除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇,与现有技术相比,本技术的制备方法简单、步骤少、成本低、反应条件温和、容易纯化、目标产物的收率高。
具体实施方式
[0025]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
[0026]
如背景技术所分析的,现有技术中存在除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇的制备方法复杂、成本较高的问题,为解决该问题,本发明提供了一种3-氯-β-亚甲基苯乙醇类化合物及其中间体各自的制备方法。
[0027]
在本技术的一种典型的实施方式中,提供了一种3-氯-β-亚甲基苯乙醇类化合物的中间体的制备方法,该制备方法包括:步骤s1,将3-氯-苯甲酰氯类化合物与氯甲烷格氏试剂进行亲核加成反应后水解,得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系;步骤s2,对中间体体系的3-氯-α,α-二甲基苯甲醇类化合物进行脱水反应,得到3-氯-β-亚甲基苯乙醇类化合物的中间体,3-氯-β-亚甲基苯乙醇类化合物的中间体为1-氯-3-(1-甲基乙烯基)苯类化合物,其中,3-氯-苯甲酰氯类化合物、3-氯-α,α-二甲基苯甲醇类化合物、1-氯-3-(1-甲基乙烯基)苯类化合物的结构式依次如下:
[0028][0029]
r选自h、c1~c
10
的烷基、c1~c
10
的烷氧基、f、cl、三氟甲基中的任意一种。
[0030]
本技术采用廉价易得的3-氯-苯甲酰氯类化合物为原料,先与氯甲烷格氏试剂进行亲核加成反应并水解得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系。3-氯-α,α-二甲基苯甲醇类化合物经脱水反应得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物。该方法步骤少、反应条件温和、成本低。并且将该制备方法得到的1-氯-3-(1-甲基乙烯基)苯类化合物进行催化氧化即可得到目标产物3-氯-β-亚甲基苯乙醇类化合物,可见,本技术仅需要经过三步反应,就可以得到除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇,与现有技术相比,本技术的制备方法简单、步骤少、成本低、反应条件温和、容易纯化、目标产物的收率高。
[0031]
在本技术的一种实施例中,上述r选自h、c1~c6的烷基、c1~c6的烷氧基中的任意一种,优选r选自h、甲基、乙基、正丙基、甲氧基、乙氧基中的任意一种,进一步地,优选r为h。
[0032]
上述不同r取代基对应不同的3-氯-苯甲酰氯类化合物,从而极大地丰富了3-氯-β-亚甲基苯乙醇类化合物的种类,且优选的上述取代基对应的3-氯-苯甲酰氯类化合物更有利于综合r取代基的电子效应和空间位阻效应,使得3-氯-苯甲酰氯类化合物的酰氯取代基具有更高的反应活性,并且最终得到的3-氯-β-亚甲基苯乙醇类化合物具有更广泛的应用。
[0033]
控制上述3-氯-苯甲酰氯类化合物与氯甲烷格氏试剂的摩尔比为1:2.2~3.0,在提高3-氯-苯甲酰氯类化合物的亲核加成反应的效率,促使3-氯-苯甲酰氯类化合物尽可能地转化为3-氯-α,α-二甲基苯甲醇类化合物的同时,又不至于对氯甲烷格氏试剂造成浪费。
[0034]
氯甲烷格氏试剂具有较强的反应活性,优选上述亲核加成反应的温度为20~35℃,优选亲核加成反应的时间为2~3小时,从而使亲核加成反应具有较高产率的基础上,尽可能地降低反应能耗并缩短反应时间。
[0035]
氯甲烷格氏试剂的制备可以参照以下步骤:
[0036]
先向1000ml的玻璃三口瓶中,加入200ml的四氢呋喃和24g(1mol)的镁屑,氮气置换充分。之后升温至50~55℃,停搅拌,向反应瓶中加入一粒碘,通入少许氯甲烷至格氏引发。确认引发控温45~55℃之间,通入氯甲烷(通入氯甲烷的量为镁屑的1.3~1.5倍),控制通入速度1~3g/min,从而控温,至镁屑反应完全,得到氯甲烷格氏试剂。
[0037]
优选上述水解在10~15wt%的盐酸溶液中进行,优选水解的温度为0~30℃,从而提高了亲核加成反应的水解效率,快速得到3-氯-α,α-二甲基苯甲醇类化合物的中间体体系。
[0038]
格氏试剂与水或者其它具有活泼h的试剂能够很轻易的进行亲核反应,优选上述亲核加成反应的溶剂选自四氢呋喃、2-甲基四氢呋喃、乙醚、甲苯中的任意一种或多种,从
而尽可能地降低溶剂对上述亲核加成反应的破坏几率。
[0039]
在本技术的一种实施例中,上述脱水反应在脱水剂作用下进行,脱水剂为3-氯-α,α-二甲基苯甲醇类化合物的1~5wt%,优选脱水剂选自甲苯-4-磺酸、蒙脱石、硫酸高铈、硫酸锌中的任意一种或多种。
[0040]
上述脱水含量及其种类有利于提高脱水反应的效率。
[0041]
在本技术的一种实施例中,上述脱水反应中还包括有抗氧化剂,抗氧化剂为3-氯-α,α-二甲基苯甲醇类化合物的0.1~0.5wt%,优选抗氧化剂选自2,6-二叔丁基-4-甲基苯酚、对叔丁基邻苯二酚、对苯二酚中的任意一种或多种。
[0042]
以上脱水反应生成的1-氯-3-(1-甲基乙烯基)苯类化合物容易聚合,以上抗氧化剂的含量及种类有利于尽可能地降低其聚合的发生几率。
[0043]
优选上述脱水反应的溶剂选自甲苯、二甲苯、环己烷、甲基环己烷、正庚烷、石油醚中的任意一种或多种,优选脱水反应的温度为80~140℃,优选脱水反应的时间为8~10h,根据所用溶剂的沸点,设置脱水反应的温度,从而在各溶剂的回流温度下提高脱水反应的效率。
[0044]
在本技术的另一种典型的实施方式中,提供了一种3-氯-β-亚甲基苯乙醇类化合物的制备方法,该制备方法包括:通过前述的制备方法得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物,将1-氯-3-(1-甲基乙烯基)苯类化合物进行催化氧化反应,得到3-氯-β-亚甲基苯乙醇类化合物,3-氯-β-亚甲基苯乙醇类化合物的结构式为:
[0045][0046]
其中,r选自h、c1~c
10
的烷基、c1~c
10
的烷氧基、f、cl、三氟甲基中的任意一种或多种。
[0047]
本技术采用廉价易得的3-氯-苯甲酰氯类化合物为原料,先与氯甲烷格氏试剂进行亲核加成反应并水解得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系。3-氯-α,α-二甲基苯甲醇类化合物经脱水反应得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物。该方法步骤少、反应条件温和、成本低。并且将该制备方法得到的1-氯-3-(1-甲基乙烯基)苯类化合物直接进行催化氧化即可得到目标产物3-氯-β-亚甲基苯乙醇类化合物,可见,本技术仅需要经过三步反应,就可以得到除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇,与现有技术相比,本技术的制备方法简单、步骤少、成本低、反应条件温和、容易纯化、目标产物的收率高。
[0048]
在本技术的一种实施例中,上述催化氧化反应在氧化剂、氧化催化剂及醇类溶剂中进行,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯类化合物的质量比为0.1~0.5:1;氧化催化剂包括过渡金属氧化物与γ-al2o3载体,过渡金属氧化物包括氧化锰、氧化钴和氧化铈,氧化锰、氧化钴、氧化铈与γ-al2o3载体的质量比为0.001~0.05:0.001~0.05:0.001~
0.01:1;氧化剂与1-氯-3-(1-甲基乙烯基)苯类化合物的摩尔比为1.2~1.5:1,优选氧化剂选自含氧气体、双氧水、次氯酸钠中的任意一种或多种,含氧气体中的氧气含量为1~50%;优选醇类溶剂选自甲醇、乙醇、异丙醇中的任意一种或多种;优选催化氧化反应的温度为20~35℃,优选催化氧化反应的时间为8~10h。
[0049]
上述氧化剂有助于将1-氯-3-(1-甲基乙烯基)苯类化合物的甲基氧化为醇,氧化催化剂的种类和含量可以促进催化氧化反应的进行,醇类溶剂有利于1-氯-3-(1-甲基乙烯基)苯类化合物、氧化催化剂、氧化剂在其中的分散,从而提高催化氧化反应体系中各成分彼此的接触,优选的催化氧化反应的温度和时间,进一步地提高了催化氧化反应的效率。
[0050]
以上氧化催化剂可以采用现有技术中已有的商业催化剂,以下提供一种氧化催化剂的制备方法,供本领域技术人员参考:
[0051]
催化剂载体的处理:将150g活性γ-al2o3微球用去离子水冲洗,除去微尘,沥干后用150g的2%的稀硝酸溶液浸泡1h,用水洗净、沥干,然后用0.5mol/l的100ml稀hcl浸泡12h,用去离子水冲洗后烘干,得到经过活化处理的γ-al2o3活性微球,将其作为氧化催化剂的载体。
[0052]
配制浸渍溶液(钴、锰、铈的摩尔比为1~10∶1~10∶0.1~1):称取2.120g的co(ch3coo)2·
4h2o、2.080g的mn(ch3coo)2·
4h2o和0.32g的cecl3·
7h2o溶解成混合溶液约100ml,加醋酸或稀盐酸调节ph=1~3。
[0053]
负载和定位:称取100g催化剂载体,在搅拌条件下,加入配置好的浸渍溶液,浸渍吸附2h后停止搅拌,静置过夜,得到粉红色球体;将该粉红色球体置于0.1~0.3mol/l的koh溶液中,间歇搅动,浸泡60min得到催化剂前驱体。
[0054]
焙烧:将得到的催化剂前驱体沥干后烘干;再将烘干后的淡蓝色球体置于马弗炉中450~480℃焙烧4~6h,自然冷却后得到蓝色球体即为负载型钴、锰、铈金属的氧化催化剂。
[0055]
以下将结合具体实施例和对比例,对本技术的有益效果进行说明。
[0056]
以下实施例和对比例中用到的氯甲烷格氏试剂的合成均参照以下步骤:
[0057]
先向1000ml的玻璃三口瓶中,加入200ml的四氢呋喃和24g(1mol)的镁屑,氮气置换充分。之后升温至55℃,停搅拌,向反应瓶中加入一粒碘,通入少许氯甲烷至格氏引发。确认引发控温50℃之间,通入氯甲烷(通入氯甲烷的量为镁屑的1.5倍),控制通入速度1g/min,从而控温至镁屑反应完全,得到氯甲烷格氏试剂。
[0058]
实施例1
[0059]
氧化催化剂的合参照以下步骤:
[0060]
催化剂载体的处理:将150g活性γ-al2o3微球用去离子水冲洗,除去微尘,沥干后用150g的2%的稀硝酸溶液浸泡1h,用水洗净、沥干,然后用0.5mol/l的100ml稀hcl浸泡12h,用去离子水冲洗后烘干,得到经过活化处理的γ-al2o3活性微球,将其作为氧化催化剂的载体。
[0061]
配制浸渍溶液(钴、锰、铈的摩尔比为10:10:1):称取2.120g的co(ch3coo)2·
4h2o、2.080g的mn(ch3coo)2·
4h2o和0.32g的cecl3·
7h2o溶解成混合溶液约100ml,加醋酸或稀盐酸调节ph=1。
[0062]
负载和定位:称取100g催化剂载体,在搅拌条件下,加入配置好的浸渍溶液,浸渍
吸附2h后停止搅拌,静置过夜,得到粉红色球体;将该粉红色球体置于0.1mol/l的koh溶液中,间歇搅动,浸泡60min得到催化剂前驱体。
[0063]
焙烧:将得到的催化剂前驱体沥干后烘干;再将烘干后的淡蓝色球体置于马弗炉中450℃焙烧4h,自然冷却后得到蓝色球体即为负载型钴、锰、铈金属的氧化催化剂。
[0064]
第一步,3-氯-α,α-二甲基苯甲醇(3c)的合成
[0065][0066]
30℃下,将氯甲烷格氏试剂(74.79g,1.00mol,2.5eq)加入反应容器内,并向反应容器内滴加280g的25wt%的3-氯-苯甲酰氯(3d)(70g,0.40mol,1eq)甲苯溶液,并控制滴加速度,使得反应液不因放热而超温;通完后保持液温30℃继续搅拌反应3小时。将反应液倒入400g的10wt%的稀盐酸中控温5~10℃进行水解,水解后继续搅拌10分钟,分液。有机层用150g的水多次水洗至中性,控温80℃以下减压浓缩至无溶剂蒸出,得到66g的3-氯-α,α-二甲基苯甲醇(3c)粗品,其收率为96.78%,其核磁数据为1hnmr(400mhz,cdcl3)δ:7.50(s,1h),7.36(d,j=8.0hz,1h),7.27(t,j=8.0hz,1h),7.22(d,j=8.0hz,1h),1.75(bs,1h),1.57(s,6h)。
[0067]
第二步,1-氯-3-(1-甲基乙烯基)苯(3b)的合成
[0068][0069]
先向500ml的玻璃三口瓶中,加入200ml的环己烷,0.7g的甲苯-4-磺酸(ptsa),0.1g的2,6-二叔丁基-4-甲基苯酚(bht)和66g(0.387mol)的3-氯-α,α-二甲基苯甲醇(3c)。氮气置换充分,升温至回流反应并在80℃下反应8小时后分出反应产生的水,得反应液。准备5℃的40g水,把反应液慢慢加到水中;一边加入一边继续搅拌和降温,并控制加入反应液的速度,使得温度保持在25℃之间。加完后,继续搅拌10分钟后静置、分液。再用40g水对有机层进行多次水洗、分液,至水层的ph值达到5~7之间后,有机层浓缩除去溶剂,即得57g的1-氯-3-(1-甲基乙烯基)苯(3b)的粗品。该粗品经减压蒸馏(液温105~115℃,顶温70~75℃,真空度30pa)除去高沸溶剂,得到53.1g的1-氯-3-(1-甲基乙烯基)苯(3b)蒸馏品,收率为90%,可直接用于下一步反应,其核磁数据为1hnmr(400mhz,cdcl3)δ:7.43(td,j=1.7,0.8hz,1h),7.34(ddd,j=6.6,2.5,1.7hz,1h),7.26-7.22(m,2h),5.38(dp,j=1.6,0.9hz,1h),5.13(p,j=1.5hz,1h),2.13(dd,j=1.5,0.8hz,3h。
[0070]
第三步,1-氯-3-(1-甲基乙烯基)苯(3b)的合成
[0071][0072]
先向500ml的玻璃三口瓶中,加入100ml的乙醇,20g氧化催化剂,50g的1-氯-3-(1-甲基乙烯基)苯(3b)蒸馏品。氮气置换充分,控温30℃,缓慢控制通入氧气含量为5%的含氧气体,含氧气体与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1.3:1。反应10小时。压滤除去氧化催化剂,滤液浓缩除去溶剂,即得54.1g的3-氯-β-亚甲基苯乙醇(1a)的粗品。该粗品减压蒸馏蒸(液温115~120℃,顶温80~85℃,真空度30pa)除去高沸溶剂,得到51.3g的3-氯-β-亚甲基苯乙醇(1a)蒸馏品,收率为93%。其核磁数据为1hnmr(400mhz,cdcl3)δ:1.61(1h,s),4.50(2h,s),5.38(1h,d,j=0.4),5.47(1h,d,j=0.4),7.33-7.39(3h,m),7.42(1h,s)。
[0073]
实施例2
[0074]
实施例2与实施例1的区别在于,第一步中,氯甲烷格氏试剂(65.82g,0.88mol,2.2eq),最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0075]
实施例3
[0076]
实施例3与实施例1的区别在于,第一步中,氯甲烷格氏试剂(89.75g,1.20mol,3.0eq),最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0077]
实施例4
[0078]
实施例4与实施例1的区别在于,第一步中,氯甲烷格氏试剂(59.83g,0.80mol,2.0eq),最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0079]
实施例5
[0080]
实施例5与实施例1的区别在于,第一步中,氯甲烷格氏试剂(104.71g,1.40mol,3.5eq),最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0081]
实施例6
[0082]
实施例6与实施例1的区别在于,第一步中,亲核加成反应的温度为35℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0083]
实施例7
[0084]
实施例7与实施例1的区别在于,第一步中,亲核加成反应的温度为20℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0085]
实施例8
[0086]
实施例8与实施例1的区别在于,第一步中,亲核加成反应的温度为40℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0087]
实施例9
[0088]
实施例9与实施例1的区别在于,第一步中,反应液倒入400g的12wt%的稀盐酸中控温5~10℃进行水解,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0089]
实施例10
[0090]
实施例10与实施例1的区别在于,第一步中,反应液倒入400g的15wt%的稀盐酸中控温5~10℃进行水解,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0091]
实施例11
[0092]
实施例11与实施例1的区别在于,第一步中,反应液倒入400g的8wt%的稀盐酸中控温5~10℃进行水解,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0093]
实施例12
[0094]
实施例12与实施例1的区别在于,第一步中,反应液倒入400g的10wt%的稀盐酸中控温0~5℃进行水解,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0095]
实施例13
[0096]
实施例13与实施例1的区别在于,第一步中,反应液倒入400g的10wt%的稀盐酸中控温25~30℃进行水解,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0097]
实施例14
[0098]
实施例14与实施例1的区别在于,第一步中,亲核加成反应的溶剂为四氢呋喃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0099]
实施例15
[0100]
实施例15与实施例1的区别在于,第二步中,1.65g的甲苯-4-磺酸(ptsa),ptsa为3-氯-α,α-二甲基苯甲醇的2.5wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0101]
实施例16
[0102]
实施例16与实施例1的区别在于,第二步中,3.3g的甲苯-4-磺酸(ptsa),ptsa为3-氯-α,α-二甲基苯甲醇的5wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0103]
实施例17
[0104]
实施例17与实施例1的区别在于,第二步中,0.528g的甲苯-4-磺酸(ptsa),ptsa为3-氯-α,α-二甲基苯甲醇的0.8wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0105]
实施例18
[0106]
实施例18与实施例1的区别在于,第二步中,3.63g的甲苯-4-磺酸(ptsa),ptsa为3-氯-α,α-二甲基苯甲醇的5.5wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0107]
实施例19
[0108]
实施例19与实施例1的区别在于,第二步中,脱水剂为硫酸高铈,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0109]
实施例20
[0110]
实施例20与实施例1的区别在于,第二步中,bht为0.066g,bht为3-氯-α,α-二甲基苯甲醇的0.1wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0111]
实施例21
[0112]
实施例21与实施例1的区别在于,第二步中,bht为0.330g,bht为3-氯-α,α-二甲基苯甲醇的0.5wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0113]
实施例22
[0114]
实施例22与实施例1的区别在于,第二步中,bht为0.053g,bht为3-氯-α,α-二甲基苯甲醇的0.08wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0115]
实施例23
[0116]
实施例23与实施例1的区别在于,第二步中,bht为0.363g,bht为3-氯-α,α-二甲基苯甲醇的0.55wt%,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0117]
实施例24
[0118]
实施例24与实施例1的区别在于,第二步中,bht为对苯二酚,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0119]
实施例25
[0120]
实施例25与实施例1的区别在于,第二步中,脱水反应的溶剂为甲苯,脱水反应的温度为110℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0121]
实施例26
[0122]
实施例26与实施例1的区别在于,第三步中,氧化催化剂为5g,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯的质量比为0.1:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0123]
实施例27
[0124]
实施例27与实施例1的区别在于,第三步中,氧化催化剂为25g,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯的质量比为0.5:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0125]
实施例28
[0126]
实施例28与实施例1的区别在于,第三步中,氧化催化剂为4g,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯的质量比为0.08:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0127]
实施例29
[0128]
实施例29与实施例1的区别在于,第三步中,氧化催化剂为30g,氧化催化剂与1-氯-3-(1-甲基乙烯基)苯的质量比为0.6:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0129]
实施例30
[0130]
实施例30与实施例1的区别在于,第三步中,氧化锰、氧化钴、氧化铈与γ-al2o3载体的质量比为0.01:0.05:0.01:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0131]
实施例31
[0132]
实施例31与实施例1的区别在于,第三步中,减少通入含氧气体量,使含氧气体与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1.2:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0133]
实施例32
[0134]
实施例32与实施例1的区别在于,第三步中,增加通入含氧气体量,使含氧气体与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1.5:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0135]
实施例33
[0136]
实施例33与实施例1的区别在于,第三步中,减少通入含氧气体量,使含氧气体与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0137]
实施例34
[0138]
实施例34与实施例1的区别在于,第三步中,增加通入含氧气体量,使含氧气体与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1.6:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0139]
实施例35
[0140]
实施例35与实施例1的区别在于,第三步中,氧化剂为次氯酸钠,次氯酸钠与1-氯-3-(1-甲基乙烯基)苯的摩尔比为1.3:1,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0141]
实施例36
[0142]
实施例36与实施例1的区别在于,第三步中,醇类溶剂为异丙醇,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0143]
实施例37
[0144]
实施例37与实施例1的区别在于,第三步中,催化氧化反应的温度为20℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0145]
实施例38
[0146]
实施例38与实施例1的区别在于,第三步中,催化氧化反应的温度为35℃,最终得到3-氯-β-亚甲基苯乙醇(1a)。
[0147]
对比例1
[0148][0149]
先向1000ml的三口瓶中加入24g(1mol)镁屑,再加入150ml的四氢呋喃,氮气置换充分,升温至50~55℃,滴入2g间溴氯苯,碘褪色后确认引发成功。控温40~45℃,滴加200ml甲苯和191.5g(1mol)间溴氯苯的混合物,滴加完毕,40~45℃下保温3小时,再降温至室温,向格氏试剂中加入9.6g(0.05mol)碘化亚铜,控温20~30℃,缓慢滴入56g(1mol)丙炔醇,滴完后升温至回流反应24小时,冷却后,滴入饱和的氯化铵溶液调ph=5,饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋干溶剂,减压精馏得到50.2g(收率:29.80%)3-氯-β-亚甲基苯乙醇(1a)。
[0150]
通过hplc分别测得上述实施例1至38中第一步反应的总收率,第一步与第二步反应的总收率,第一步、第二步与第三步反应的总收率,并将测试结果列于表1。
[0151]
表1
[0152]
[0153]
[0154][0155]
实施例1与实施例23的对照说明,脱水剂的用量增加对第二步反应收率的影响不大,但会造成试剂的浪费而增加成本
[0156]
实施例1、实施例29、实施例34的对照说明,第三步反应中,氧化催化剂、氧化剂的用量增加对第三步反应收率的影响不大,但会造成试剂的浪费而增加成本。
[0157]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
[0158]
本技术采用廉价易得的3-氯-苯甲酰氯类化合物为原料,先与氯甲烷格氏试剂进行亲核加成反应并水解得到包括3-氯-α,α-二甲基苯甲醇类化合物的中间体体系。3-氯-α,α-二甲基苯甲醇类化合物经脱水反应得到3-氯-β-亚甲基苯乙醇类化合物的中间体,即1-氯-3-(1-甲基乙烯基)苯类化合物。该方法步骤少、反应条件温和、成本低。并且将该制备方法得到的1-氯-3-(1-甲基乙烯基)苯类化合物进行催化氧化即可得到目标产物3-氯-β-亚甲基苯乙醇类化合物,可见,本技术仅需要经过三步反应,就可以得到除草剂茚草酮的主要原料3-氯-β-亚甲基苯乙醇,与现有技术相比,本技术的制备方法简单、步骤少、成本低、反应条件温和、容易纯化、目标产物的收率高。
[0159]
以上仅所述为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1