一种神经激肽-1拮抗剂的晶型及其制备方法与流程
1.本公开涉及一种神经激肽-1拮抗剂的晶型,具体地涉及式ii化合物的a晶型及制备方法。
背景技术:
2.速激肽是神经激肽受体的肽配体。神经激肽受体,比如nk1,nk2和nk3,与各种生物过程有关。可在哺乳动物的神经和循环系统以及在周围组织中发现它们。因此,这类受体的调节已被研究用于潜在地治疗或预防哺乳动物的各种生理失调、病症或疾病。
3.另一方面,药物溶血性是药物进入人体后免疫因素导致的红细胞大量破坏引起的,临床上出现了贫血、黄疸、酱油和尿液等溶血现象。药物性溶血性贫血可分为以下三种类型:(1)药物性免疫,导致抗体介导的溶血性反应;(2)药物作用于具有遗传酶缺陷的红细胞(例如g6pd缺陷);(3)药物对异常血红蛋白的溶血反应。治疗这种疾病的关键在于停止使用相关药物,控制溶血的发生,以防止并发症的发生。
4.pct/cn2020/098460提供了一种新的对治疗各种生理失调、病征和疾病有效而副作用最小的nk1拮抗剂前药化合物(式ii),该化合物具有较好的药学活性。
[0005][0006]
药用的活性成分的晶型结构往往影响到该药物的化学稳定性,结晶条件及储存条件的不同有可能导致化合物的晶型结构的变化,有时还会伴随着产生其他形态的晶型。一般来说,无定形的药物产品没有规则的晶型结构,往往具有其它缺陷,比如产物稳定性较差,析晶较细,过滤较难,易结块,流动性差等。因此,改善式ii所示化合物的各方面性质是很有必要的,我们需要深入研究找到晶型纯度较高并且具备良好化学稳定的新晶型。
技术实现要素:
[0007]
本公开提供了一种式ii所示化合物的a晶型,其具备良好的稳定性,可更好地应用于临床。
[0008][0009]
本公开提供的式ii所示化合物的a晶型,其x-射线粉末衍射图谱在2θ角为10.336、12.530、17.987、18.487、20.937处有特征峰。
[0010]
本公开提供的式ii所示化合物的a晶型,其x-射线粉末衍射图谱在2θ角为10.336、11.705、12.530、16.848、17.987、18.487、19.840、20.937、22.865、23.580处有特征峰。
[0011]
本公开提供的式ii所示化合物的a晶型,其x-射线粉末衍射图谱在2θ角为8.367、10.336、11.150、11.705、12.530、13.306、14.236、15.252、16.330、16.848、17.459、17.987、18.487、19.294、19.840、20.937、21.963、22.285、22.865、23.580、24.812、25.961、26.798、27.597、28.761、29.784、31.737、32.470、35.717、37.977、42.828、46.938处有特征峰。
[0012]
本公开提供的式ii所示化合物的a晶型,其x-射线粉末衍射图谱如图2所示。
[0013]
本公开提供的式ii所示化合物的a晶型,其中所述2θ角的误差范围为
±
0.2。
[0014]
本公开进一步提供一种制备式ii所示化合物的a晶型的方法,所述方法包括:将式ii所示化合物与适量的溶剂混合打浆,分离固体,所述溶剂为醚类,优选乙醚。
[0015]
通过x-射线粉末衍射图谱(xrpd)、差示扫描量热分析(dsc)对本公开所得到晶型进行结构测定、晶型研究。
[0016]
本公开中晶型的析晶方法是常规的,例如挥发析晶、降温析晶或室温下析晶。
[0017]
本公开晶型制备方法中所用的起始原料可以是任意形式的式ii所示化合物,具体形式包括但不限于:无定形、任意晶型、水合物、溶剂合物等。
[0018]
本公开进一步提供一种药物组合物,包含式ii所示化合物的a晶型,以及一种或多种药学上可接受的载体或赋形剂。
[0019]
本公开进一步提供一种药物组合物,其通过式ii所示化合物的a晶型,与一种或多种药学上可接受的载体或赋形剂制备得到。
[0020]
本公开进一步提供一种药物组合物的制备方法,包括将式ii所示化合物的a晶型与一种或多种药学上可接受的载体或赋形剂混合的步骤。
[0021]
本公开进一步提供前述的式ii所示化合物的a晶型、或由前述方法制备得到的组合物在制备治疗患者中的生理失调、病症或疾病的药物中的用途,其中生理失调、病症或疾病优选呼吸道疾病、咳嗽、炎性疾病、皮肤障碍、眼科障碍、抑郁症、焦虑、恐怖症、双向障碍、酒精依赖、对神经起显著作用的物质滥用、癫痫、伤害感受、精神病、精神分裂症、阿尔茨海默氏病、与aids有关的痴呆、towne
′
s疾病、与紧张有关的障碍、强迫性/强制性障碍、bulemia、神经性厌食症、疯狂进食、狂躁、经前期综合征、胃肠机能紊乱、动脉粥样硬化、纤维化障碍、肥胖、ii型糖尿病、头痛、神经性疼痛、动作后疼痛、慢性疼痛综合症、膀胱障碍、泌尿生殖器障碍或呕吐或恶心。
[0022]
在本技术的说明书和权利要求书中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。然而,为了更好地理解本公开,下面提供了部分相关术语的定义和解释。另外,当本技术所提供的术语的定义和解释与本领域技术人员所通常理解的含义不一致时,以本技术所提供的术语的定义和解释为准。
[0023]
本公开所述的“x-射线粉末衍射图谱或xrpd”是指根据布拉格公式2d sinθ=nλ(式中,λ为x射线的波长,衍射的级数n为任何正整数,一般取一级衍射峰,n=1),当x射线以掠角θ(入射角的余角,又称为布拉格角)入射到晶体或部分晶体样品的某一具有d点阵平面间距的原子面上时,就能满足布拉格方程,从而测得了这组x射线粉末衍射图。
[0024]
本公开所述的“x-射线粉末衍射图谱或xrpd”是通过在x-射线粉末衍射仪中使用cu-kα辐射得到的图谱。
[0025]
本公开所述的“差示扫描量热分析或dsc”是指在样品升温或恒温过程中,测量样品与参考物之间的温度差、热流差,以表征所有与热效应有关的物理变化和化学变化,得到样品的相变信息。
[0026]
本公开所述的“热重分析或tga”是指在程序控制温度下,连续测量样品的质量随温度或时间的变化。
[0027]
本公开所述的“2θ或2θ角度”是指衍射角,θ为布拉格角,单位为
°
或度,2θ的误差范围为
±
0.3或
±
0.2或
±
0.1。
[0028]
本公开所述的“晶面间距或晶面间距(d值)”是指空间点阵选择3个不相平行的连结相邻两个点阵点的单位矢量a,b,c,它们将点阵划分成并置的平行六面体单位,称为晶面间距。空间点阵按照确定的平行六面体单位连线划分,获得一套直线网格,称为空间格子或晶格。点阵和晶格是分别用几何的点和线反映晶体结构的周期性,不同的晶面,其面间距(即相邻的两个平行晶面之间的距离)各不相同;单位为或埃。
附图说明
[0029]
图1为式ii所示化合物的无定形的xrpd图谱;
[0030]
图2为式ii所示化合物a晶型的xrpd图谱;
[0031]
图3为式ii所示化合物a晶型的dsc图谱;
[0032]
图4为式ii所示化合物a晶型的tga图谱。
具体实施方式
[0033]
以下将结合实施例更详细地解释本公开,本公开的实施例仅用于说明本公开的技术方案,并非限定本公开的实质和范围。
[0034]
试验所用仪器的测试条件:
[0035]
1、差示扫描量热仪(differential scanning calorimeter,dsc)
[0036]
仪器型号:mettler toledo dsc 3+
[0037]
吹扫气:氮气,50ml/min
[0038]
升温速率:10.0℃/min
[0039]
温度范围:25-300℃
[0040]
2、x-射线粉末衍射仪(x-ray powder diffraction,xrpd)
[0041]
仪器型号:bruker d8 discover
[0042]
射线:单色cu-kα射线(cu-kα1波长为cu-kα2波长为cu-kα波长取kα1与kα2的加权平均值)
[0043]
扫描方式:θ/2θ,扫描范围:3-50
°
[0044]
电压:40kv,电流:40ma
[0045]
3、热重分析仪(thermogravimetric analysis,tga)
[0046]
仪器型号:mettler toledo tga 2
[0047]
吹扫气:氮气,50ml/min
[0048]
升温速率:10.0℃/min
[0049]
温度范围:25-350℃
[0050]
化合物的结构是通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(δ)以10-6
(ppm)的单位给出。nmr的测定是用bruker avance-400核磁仪,测定溶剂为氘代二甲基亚砜(dmso-d6)、氘代氯仿(cdcl3)、氘代甲醇(cd3od),内标为四甲基硅烷(tms)。
[0051]
ms的测定用finnigan lcqad(esi)质谱仪(生产商:thermo,型号:finnigan lcq advantage max)。
[0052]
高效液相色谱法(hplc)分析使用agilent hplc 1200dad、agilent hplc 1200vwd和waters hplc e2695-2489高压液相色谱仪。
[0053]
手性hplc分析测定使用agilent 1260dad高效液相色谱仪。
[0054]
高效液相制备使用waters 2767、waters 2767-sq detecor2、shimadzu lc-20ap和gilson-281制备型色谱仪。
[0055]
手性制备使用shimadzu lc-20ap制备型色谱仪。
[0056]
combiflash快速制备仪使用combiflash rf200(teledyne isco)。
[0057]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.15mm~0.2mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
[0058]
硅胶柱色谱法一般使用烟台黄海硅胶200~300目硅胶为载体。
[0059]
本公开的已知的起始原料可以采用或按照本领域已知的方法来合成,或可购买自abcr gmbh&co.kg,acros organics,aldrich chemical company,韶远化学科技(accela chembio inc)、达瑞化学品等公司。
[0060]
实施例中无特殊说明,反应能够均在氩气氛或氮气氛下进行。
[0061]
氩气氛或氮气氛是指反应瓶连接一个约1l容积的氩气或氮气气球。
[0062]
氢气氛是指反应瓶连接一个约1l容积的氢气气球。
[0063]
加压氢化反应使用parr 3916ekx型氢化仪和清蓝ql-500型氢气发生器或hc2-ss型氢化仪。
[0064]
氢化反应通常抽真空,充入氢气,反复操作3次。
[0065]
微波反应使用cem discover-s 908860型微波反应器。
[0066]
实施例中无特殊说明,溶液是指水溶液。
[0067]
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
[0068]
实施例中的反应进程的监测采用薄层色谱法(tlc),反应所使用的展开剂,纯化化
合物采用的柱层析的洗脱剂的体系和薄层色谱法的展开剂体系包括:a:正己烷/乙酸乙酯体系,b:二氯甲烷/甲醇体系,溶剂的体积比根据化合物的极性不同而进行调节,也可以加入少量的三乙胺和醋酸等碱性或酸性试剂进行调节。
[0069]
实施例1:式ii化合物的制备
[0070][0071]
第一步:
[0072][0073]
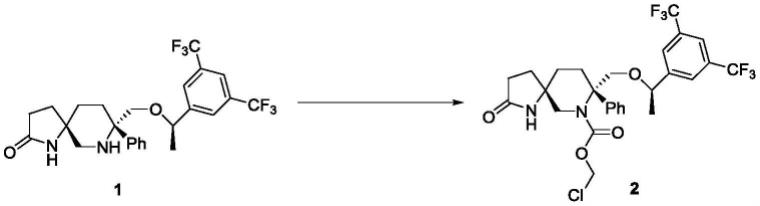
在n2保护下,于100ml三口瓶中称取化合物1(2.43g,4.86mmol,1eq)溶于二氯甲烷(36ml)中,加入二异丙基乙胺(5g,38.76mmol,8eq),冷却到-30℃,加入三甲基氯硅烷(1.36g,12.52mmol,2.6eq),室温下搅拌2h。再冷却到-25℃,滴加氯甲酸氯甲酯(0.77g,6mmol,1.23eq)的二氯甲烷溶液,控温-20℃~-5℃搅拌至反应完毕,将反应液倒入冰水中,分液,二氯甲烷萃取,加入水和1n的盐酸溶液,分液,再依次用食盐水、饱和碳酸氢钠水溶液和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩,得到3.0g黄色胶状物,收率104%。
[0074]
第二步:
[0075][0076]
在n2保护下,于500ml三口瓶中加入化合物2(2.8g,4.53mmol,1eq),四丁基碘化铵(1.68g,4.55mmol,1eq),磷酸二叔丁酯钾盐(5.63g,22.67mmol,5eq)和二氧六环(84ml),加热至55℃下搅拌4h。反应液降温,倒入乙酸乙酯和水中,分液,用乙酸乙酯萃取,用亚硫酸钠的水溶液洗涤,再依次用水、食盐水洗涤,无水硫酸钠干燥,过滤,浓缩得到3.73g黄色泡沫,收率107%。
[0077]
第三步:
[0078][0079]
在n2保护下,于500ml单口瓶中加入化合物3(6.65g,8.67mmol,1eq)溶于二氯甲烷(200ml)中,冰水冷却下缓慢加入三氟乙酸(9.89g,86.7mmol,10.0eq),搅拌至反应完毕,浓缩,得到2.29g油状物,经反相硅胶柱(c18)纯化(a溶液:20mmol nh4hco3水溶液,b溶液:乙腈)纯化,再用1m磷酸调节ph=1~2,用二氯甲烷萃取,饱和盐水洗涤,无水硫酸钠干燥,过滤,浓缩得目标产物2.7g。
[0080]1h-nmr(400mhz,cd3od):δ(ppm)7.89(s,2h),7.86(s,1h),7.41-7.27(m,5h),5.66(d,j=12hz,1h),5.50-5.47(m,1h),4.60(d,j=8hz,1h),4.20-3.88(m,3h),2.51-2.10(m,5h),1.86-1.66(m,3h),1.44-1.31(m,4h).
[0081]
经x-射线粉末衍射检测,化合物4(即式ii所示化合物)为无定形,xrpd谱图如图1。
[0082]
测试例1:溶解度
[0083]
1.1配制试剂
[0084]
试剂:nah2po4·
2h2o
[0085]
1.2配制方法
[0086]
按100ml规格配制如下:
[0087]
ph=3.0:磷酸盐缓冲溶液:100ml 20mmol/l nah2po4,0.1m h3po4调节ph至3.0。
[0088]
ph=4.0:磷酸盐缓冲溶液:100ml 20mmol/l nah2po4,0.1m h3po4调节ph至4.0。
[0089]
ph=7.0:超纯水
[0090]
ph=9.0:磷酸盐缓冲溶液:100ml 20mmol/l na2hpo4,0.1m naoh溶液调节ph至9.0
[0091]
1.3测试方法
[0092]
精确称取适量实施例1化合物,少量多次加入溶液搅拌等待化合物溶解,测定溶液中化合物含量。数据见表1。
[0093]
表1
[0094]
ph溶解度饱和溶解度7.426mg/ml19.8mg/ml9.028mg/ml21.4mg/ml
[0095]
测试例2:溶血作用
[0096]
红血球(rbc)随机地从兔颈静脉或耳中央动脉采取(edta全血)10ml,放入玻璃珠的三角烧瓶中振摇10分钟,除去纤维蛋白原,使成脱纤血液。加入氯化钠注射液约10倍量,摇匀,1500转/分钟离心10分钟,除去上清液,沉淀的红细胞再用氯化钠注射液按上述方法
洗涤3次,至上清液不显红色。将所得红细胞用氯化钠注射液配成2%(v/v)的混悬液,以备用。
[0097]
取实施例1化合物溶于pbs(ph 7.4或ph 5),过滤,配制0.4mg/ml、0.8mg/ml、1.2mg/ml、1.6mg/ml和2mg/ml,以备用。
[0098]
取一定量的实施例1化合物溶液加入上述血红蛋白在上清液中测试。
[0099]
若试管中的溶液呈澄明红色,管底无细胞残留或有少量红细胞残留,表明有溶血发生;如红细胞全部下沉,上清液体无色澄明,表明无溶血发生。若溶液中有棕红色或红棕色絮状沉淀,轻轻倒转3-5次仍不分散,表明可能有红细胞凝聚发生;应进一步置显微镜下观察,如可见红细胞聚集为凝聚。本公开提供的化合物采用该方法测定溶血作用。
[0100]
结论:实施例1化合物在浓度高至2mg/ml都没有出现溶血作用。
[0101]
测试例3:罗拉匹坦乳剂溶血性作用
[0102]
参照cn102573475中方法制备罗拉匹坦乳剂(处方为:4.4%聚乙二醇-15羟基硬脂酸酯、1.1%中链甘油三酸酯和0.66%大豆油),用pbs配制0.18mg/ml、0.09mg/ml、0.045mg/ml、0.023mg/ml、0.011mg/ml、0.056mg/ml和0.028mg/ml,以备用。
[0103]
参照测试例2中方法测定溶血作用。
[0104]
结论:所有浓度的罗拉匹坦乳剂均存在溶血作用。
[0105]
测试例4:食蟹猴中的药代动力学测试
[0106]
以食蟹猴为受试动物,应用lc/ms/ms法测定了注射给予参照实施例1方法制备的化合物后不同时刻血浆中的药物浓度。研究该化合物在食蟹猴体内的药代动力学行为,评价其药动学特征。
[0107]
药物配制
[0108]
称取一定量待测化合物,用20mmol/l磷酸二氢钠配制成ph=4.0溶液,以备用。
[0109]
1.1给药
[0110]
静脉滴注,推注时间约30min,给药剂量3.54mg/kg,给药浓度2mg/ml,给药体积5ml/kg。
[0111]
1.2操作
[0112]
给药前及给药结束后5min、0.25h、0.5h、1h、2h、4h、6h、8h、10h和24h,经股静脉采血,每个样品采集约0.6ml,肝素钠抗凝,采集后马上放置冰上。血液样本采集后放置于标记好的离心管中,离心分离血浆(离心条件:离心力2200g,离心10min,2-8℃)。
[0113]
应用lc/ms/ms测定血浆样品中的实施例1化合物和罗拉匹坦的含量。
[0114]
1.3药代动力学参数结果
[0115]
表2
[0116]
[0117]
注:a实施例中化合物在食蟹猴体内药代动力参数,b实施例化合物代谢为罗拉匹坦在食蟹猴体内药代动力参数。
[0118]
结论:该化合物在食蟹猴体内药代动力学研究中,在食蟹猴绝大部分迅速转化为活性代谢产物罗拉匹坦,其具有良好的药代动力学性质。
[0119]
实施例2:式ii化合物a晶型的制备
[0120]
称取约100mg式ii所示化合物,加入1ml乙醚中,室温下搅拌溶清,打浆析出固体,离心后真空干燥得到式ii所示化合物的a晶型,其特征峰位置如表3所示:
[0121]
表3:a晶型的xrd特征峰位置
[0122]
[0123][0124]
实施例3:晶型a的稳定性考察
[0125]
将式ii化合物a晶型放置4℃条件下考察其稳定性。
[0126]
表4
[0127][0128]
结果显示:a晶型在4℃稳定性条件下放置3个月的物理、化学稳定性好。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1