一种星型可溶性共轭聚合物及其制备方法和应用
1.本发明属于有机无机纳米杂化材料和有机导电聚合物技术领域,尤其涉及一种星型可溶性共轭聚合物及制备方法和其应用。
背景技术:
2.近来,众所周知,聚合物的拓扑结构直接影响其化学和物理性能。设计和研究具有不同拓扑结构的聚合物功能材料是高分子材料领域的重要方向。随着聚合技术和方法的不断发展,许多具有复杂结构的非线型聚合物受到了越来越多的关注。近年来,同时包含柔性非共轭链段和刚性共轭链段的结构明确的新型复合高分子材料以其在结构和相取向方面的差异性使其拥有远比共轭单元均聚物材料更多的应用范围和关注度。按链段排布方式不同,可以将此类聚合物分为线型和枝化结构两类,如星型、树枝型、梳型等结构。基于在电化学方面的应用,与线型结构聚合物相比,枝化结构的复合高分子聚合物拥有两种结构和性质迥异的链段从而具有两个均聚物无法企及的结构优势,拥有更多的柔性链段和共轭链段,不仅能够克服小分子共轭材料机械强度弱的缺点,同时有利于不同导电相区之间电子的跃迁效应,并且能够更好的稳定一维碳材料,在光电材料的制备领域具有很强的应用前景。
3.目前已有多个利用fe
3+
氧化聚合的方式在含端基噻吩的星型聚合物先驱体上生长共轭链的报道,但是制备共轭聚合物的催化剂和反应单体在同一个相内,使反应单体容易作为一个交联剂将不同的共轭链交联在一起,破坏了设想的分子结构也降低了产物的溶解性。从而只有一些生成的含低聚噻吩链的最终产物仍可以维持之前的星型结构,而随着噻吩链段的生长,链间的偶联反应不仅会破坏聚合物的溶解性而且会影响最终产物的分子量分布。
技术实现要素:
4.本发明的第一个目的在于提供一种星型可溶性共轭聚合物。
5.本发明的第二个目的在于提供一种星型可溶性共轭聚合物的制备方法,可使产物保持良好的溶解性,并具有分子量可控的效果。
6.本发明的再一目的是提供星型可溶性共轭聚合物的应用。
7.具体地,本发明涉及以下的技术方案:
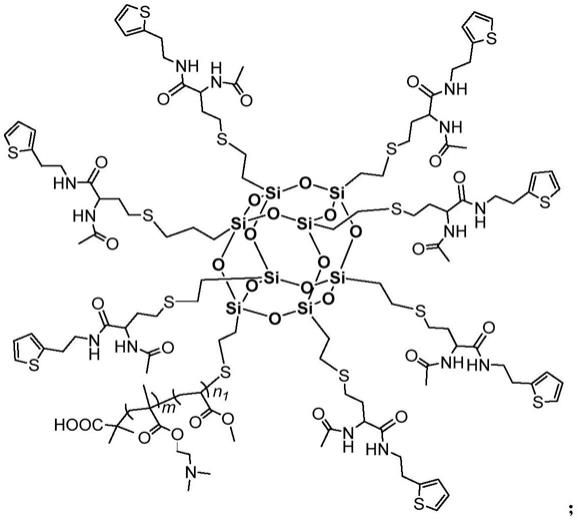
8.一种星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot],具有如下式i所述结构通式:
[0009][0010]
其中,50≤m≤100;1≤n1≤10;5≤n2≤20。
[0011]
本发明的星型可溶性共轭聚合物含有数量众多的共轭链段,使其表现出良好的导电性以及溶解性。
[0012]
在本发明的一些实例中,所述星型可溶性共轭聚合物的质均分子量为25000~45000da。
[0013]
一种星型可溶性共轭聚合物的制备方法,包括如下步骤:
[0014]
以含有星型杂化聚合物[csq-(pdma-b-pma)-th]和fe
3+
的水溶液为水相,将3,4-乙撑二氧噻吩(edot)/正丁醇溶液覆盖在所述水相上,进行界面反应,得到所述星型可溶性共轭聚合物;
[0015]
所述星型杂化聚合物的结构通式为:
[0016][0017]
所述星型可溶性共轭聚合物的结构通式为:
[0018][0019]
其中,50≤m≤100;1≤n1≤10;5≤n2≤20。
[0020]
本发明的制备方法利用一种改进的界面合成方案,在fe
3+
催化下从倍半硅氧烷基含多个噻吩活性基团的星型杂化聚合物[csq-(pdma-b-pma)-th]的端噻吩基团开始生长聚3,4-乙撑二氧噻吩(edot)链段,形成倍半硅氧烷基含多条共轭链段的星型可溶性共轭聚合物(csq-(pdma-b-pma)-pedot),合成过程中可减少反应单体将不同的共轭链交联在一起,可成功得到设想的分子结构并保持产物的溶解性,而且可有效控制产物的分子量分布。同
时,现有技术一般需要采用含有柔性链段的噻吩单体才能实现聚合物的可溶性,而本发明使用不含柔性链段的噻吩单体合成聚噻吩链段即可制得可溶性的聚合物,具有单体应用广泛、结构性质可调、经济高效的特点。
[0021]
在本发明的一些实例中,所述fe
3+
与星型杂化聚合物的摩尔比为1:5~15,优选1:5~10,更优选1:8~9。
[0022]
在本发明的一些实例中,所述水相由星型杂化聚合物的盐酸溶液与fe
3+
水溶液混合而得。
[0023]
在本发明的一些实例中,所述星型杂化聚合物的盐酸溶液由星型杂化聚合物溶于所述盐酸中得到,所述盐酸的浓度为0.1~5mol/l,优选0.5~3mol/l,更优选0.5~1.5mol/l。所述星型杂化聚合物与盐酸的比例为1g:10~100ml,优选1g:20~80ml,更优选1g:20~50ml;或者,所述星型杂化聚合物与盐酸的比例为1mmol:1~5l,优选1mmol:2~3l。
[0024]
在本发明的一些实例中,所述fe
3+
水溶液的浓度为0.05~1mol/l,优选0.08~0.5mol/l,更优选0.1~0.2mol/l。
[0025]
在本发明的一些实例中,所述fe
3+
水溶液与星型杂化聚合物的盐酸溶液的体积比为1:0.5~5:优选1:1~5,更优选1:2~3。
[0026]
在本发明的一些实例中,所述3,4-乙撑二氧噻吩/正丁醇溶液的浓度为0.005~0.5mol/l,优选0.01~0.1mol/l,更优选0.01~0.03mol/l。实际操作中,可将3,4-乙撑二氧噻吩、正丁醇按质量体积比为0.001~0.005g:1ml,优选0.002~0.003g:1ml进行配比。
[0027]
在本发明的一些实例中,所述水相与3,4-乙撑二氧噻吩/正丁醇溶液的体积比为0.1~0.5:1,优选0.3~0.4:1。
[0028]
在本发明的一些实例中,所述星型杂化聚合物和正丁醇的比例为0.005~0.02g:1ml,优选0.008~0.012g:1ml。
[0029]
在本发明的一些实例中,所述界面反应在避光条件下进行。
[0030]
在本发明的一些实例中,所述界面反应的温度为10~50℃,优选20~30℃。界面反应的时间为10~40小时,优选24~36小时。
[0031]
在本发明的一些实例中,所述界面反应结束后,还包括纯化的步骤,所述纯化的步骤包括:取界面反应结束后的下层水相,进行离心;取离心后的上层清液;将上层清液与nahco3/thf混合液混合,搅拌至无气泡溢出,静置;取静置后的上层清液进行浓缩,浓缩后在冰石油醚中沉淀,取出沉淀;将沉淀干燥,得到星型可溶性共轭聚合物。
[0032]
其中,nahco3/thf混合液为nahco3水溶液与四氢呋喃的混合液。nahco3水溶液的浓度为0.1~2mol/l,优选0.5~1.5mol/l。nahco3水溶液和四氢呋喃的体积比为1:1~5,优选1:2~4。nahco3/thf混合液与3,4-乙撑二氧噻吩/正丁醇溶液的体积比为1~2:1,优选1.2~1.6:1。石油醚与3,4-乙撑二氧噻吩/正丁醇溶液的体积比为5~30:1,优选10~20:1。
[0033]
在本发明的一些实例中,所述星型杂化聚合物的制备方法包括如下步骤:
[0034]
1)使dl-n-乙酰高半胱氨酸硫内酯(化合物1)和2-噻吩乙胺(化合物2)进行反应,得到化合物3;使化合物3与八乙烯基笼型倍半硅氧烷(csq,化合物4)反应,得到含多个端噻吩基团的八面体倍半硅氧烷(csq-th,化合物5);
[0035]
2)使聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma,化合物6)脱硫得到化合物7;使所述化合物5与化合物7反应得到化合物8所示的星型杂化聚合物。
[0036]
星型杂化聚合物的合成路线如下:
[0037][0038]
在本发明的一些实例中,所述化合物1与化合物2的反应在催化剂存在下进行,所述催化剂包括4-二甲氨基吡啶、三丁基膦、三苯基膦中的至少一种。
[0039]
在本发明的一些实例中,所述化合物1与化合物2的摩尔比为1~1.5:1;所述化合物2与化合物4的摩尔比为1:0.1~0.15。
[0040]
在本发明的一些实例中,所述化合物1与化合物2的反应在有机溶剂中进行,所述dl-n-乙酰高半胱氨酸硫内酯和有机溶剂的比例为0.1~0.3g:1ml。所述催化剂和有机溶剂
的比例为0.003~0.005g:1ml。所述2-噻吩乙胺和有机溶剂的比例为0.03~0.05g:1ml。
[0041]
在本发明的一些实例中,所述化合物3与化合物4的反应具体为,将化合物4的有机溶液加入到化合物1与化合物2反应结束后得到的含化合物3的混合物中,进行反应。其中,化合物4的有机溶液为化合物4溶于有机溶剂中得到的溶液,该有机溶剂优选与化合物1和化合物2的反应体系中的有机溶剂相同。所述化合物4的有机溶液浓度为0.1~0.5mmol/ml,优选0.2~0.3mmol/ml,由化合物4和有机溶剂体积比0.05~1:1,优选0.2~0.8:1进行配比得到。
[0042]
在本发明的一些实例中,所述化合物1与化合物2的反应,以及化合物3与化合物4的反应均在保护气氛中进行。
[0043]
在本发明的一些实例中,所述化合物1与化合物2的反应时间,以及化合物3与化合物4的反应时间独立为8~20小时。优选地,化合物1与化合物2的反应时间优选为8~12小时;化合物3与化合物4的反应时间优选为10~16小时。优选地,化合物1与化合物2的反应时间:化合物3与化合物4的反应时间为1:1~2,优选1:1~1.8。
[0044]
化合物1与化合物2的反应温度,以及化合物3与化合物4的反应温度独立为30~50℃,两步反应的温度优选相同。
[0045]
在本发明该的一些实例中,所述化合物3与化合物4反应结束后,可通过减压抽走溶剂、水洗、二氯甲烷溶解、分子筛干燥、浓缩、层析柱分离、真空干燥等步骤,获得纯化的目标产物。其中真空干燥温度为25~30℃,真空干燥时间约20~30小时。
[0046]
在本发明的一些实例中,步骤2)中,所述化合物6脱硫形成化合物7的步骤具体为:将化合物6溶于有机溶剂,加入亲核催化剂和还原剂,反应得到化合物7。反应在保护氛围下进行。
[0047]
在本发明的一些实例中,所述亲核催化剂包括三丁基膦、三苯基膦中的至少一种。所述还原剂包括丙胺、乙胺、己胺中的至少一种。
[0048]
在本发明的一些实例中,所述化合物6与亲核催化剂、还原剂的摩尔比为1:5~15:5~15,优选1:10~15:10~15。化合物6和有机溶剂按质量体积比为0.1~0.3g:1ml进行配比;亲核催化剂和有机溶剂按体积比为0.5~1.5:1进行配比;还原剂和有机溶剂按体积比为0.2~0.6:1进行配比。
[0049]
在本发明的一些实例中,所述化合物6与亲核催化剂、还原剂反应完成后,将有机溶剂和未反应的还原剂去除,然后加入化合物5/三乙胺/有机溶剂,进行反应,得到化合物8所示的星型杂化聚合物。
[0050]
化合物5/三乙胺/有机溶剂和溶解化合物6的有机溶剂按体积比为0.8~1.2:1进行配比。化合物5/三乙胺/有机溶剂中,化合物5和有机溶剂按质量体积比为0.002~0.006g:1ml进行配比,三乙胺和有机溶剂按体积比为0.01~0.02:1进行配比。
[0051]
在本发明的一些实例中,步骤2)中,化合物6脱硫形成化合物7的反应时间为3~5小时。化合物5与化合物7的反应时间为12~36小时。步骤2)的两步反应温度独立为30~50℃。
[0052]
在本发明的一些实例中,所述化合物6的制备方法为:
[0053]
s1:在引发剂作用下,使甲基丙烯酸n,n-二甲基氨基乙酯(dma,化合物a)、s-十二烷基-s
’‑
(r,r
’‑
二甲基-r
”‑
乙酸)三硫代碳酸盐(dttc,化合物b)进行反应,得到聚甲基丙
烯酸n,n-二甲基氨基乙酯(pdma,化合物c);
[0054]
s2:在引发剂作用下,使所述化合物c与丙烯酸甲酯(ma,化合物d)进行聚合反应得到化合物6。
[0055]
化合物6的合成路线如下:
[0056][0057]
在本发明的一些实例中,所述化合物a与化合物b的反应温度,以及化合物c与化合物d的反应温度独立为65~75℃,反应时间独立为2~6小时。
[0058]
在本发明的一些实例中,步骤s1中,所述化合物a与化合物b的摩尔比为0.1~0.5:1。在实际操作中,可将化合物a和化合物b溶解于有机溶剂中,然后进行反应。化合物b和有机溶剂按质量体积比为0.03~0.05g:1ml进行配比;化合物a和有机溶剂按质量体积比为1.2~1.8g:1ml进行配比;引发剂和有机溶剂按质量体积比为0.0014~0.0018g:1ml进行配比。
[0059]
在本发明的一些实例中,步骤s2中,所述化合物c与化合物d的摩尔比为15~30:1。在实际操作中,可将化合物c与化合物d溶解于有机溶剂中,然后进行反应。化合物c和有机溶剂按质量体积比为0.2~0.4g:1ml进行配比;化合物d和有机溶剂按质量体积比为0.05~0.08g:1ml进行配比;引发剂和有机溶剂按质量体积比为0.0004~0.0008g:1ml进行配比。
[0060]
在本发明的一些实例中,步骤s1和s2中,达到化合物a与化合物b所需反应时间后,或者达到化合物c与化合物d所需反应时间后,通过对反应体系进行降温(至-78℃~0℃)来淬灭反应,然后将反应液滴入冰石油醚中沉淀,对沉淀进行干燥得到目标产物。石油醚和有机溶剂按体积比为10~20:1进行配比。
[0061]
在本发明的一些实例中,本发明对所述引发剂不作特殊的限定,可采用本领域通用的聚合反应用引发剂,例如偶氮二异丁腈(aibn)。
[0062]
在本发明的一些实例中,本发明对各步骤涉及的有机溶剂不作特殊的限定,能够溶解反应物且不参与反应的有机溶剂均可使用。作为示例,本发明各步骤采用的有机溶剂独立地包括二氧六环、四氢呋喃、甲苯中的至少一种。
[0063]
上述星型可溶性共轭聚合物的应用,应用于有机光电材料制备技术领域。
[0064]
相对于现有技术,本发明具有如下有益效果:
[0065]
(1)本发明的方法可以通过聚合时间的控制来调节柔性部分链段的分子量,制备的星型可溶性共轭聚合物具有结构可控,分子量可控,分子量分布窄等特点。
[0066]
(2)本发明的反应条件温和,产物得率较高(60%以上),且制备的产物结构明确,提纯方便利于表征。
[0067]
(3)现有技术一般需要采用含有柔性链段的噻吩单体才能实现聚合物的可溶性,而本发明使用不含柔性链段的噻吩单体合成聚噻吩链段即可制得可溶性的聚合物,具有单体应用广泛、结构性质可调、经济高效的特点。
[0068]
(4)本发明用于设计制备含多个共轭链段的星型结构聚合物,能够提高聚合物内共轭部分的组成,制备出可溶且电化学性能好的导电聚合物,具有良好的应用前景。
附图说明
[0069]
图1为实施例1的csq-th的1h-nmr图谱;
[0070]
图2为实施例1中csq-(pdma-b-pma)-pedot的1h-nmr图谱;
[0071]
图3为实施例1中csq-(pdma-b-pma)-pedot的红外图谱;
[0072]
图4为实施例1中csq-(pdma-b-pma)-pedot的循环伏安曲线图。
[0073]
图5为实施例1中csq-(pdma-b-pma)-pedot溶解在四氢呋喃中的照片。
具体实施方式
[0074]
本发明结合了“grafting-onto”和“grafting-from”这两种制备星型聚合物的方式来合成倍半硅氧烷基星型可溶性共轭聚合物。首先利用环硫亲核开环-点击反应将多个噻吩基团连接到八乙烯基笼型倍半硅氧烷(csq)上,得到一组含多个端噻吩基团的八面体倍半硅氧烷(csq-th),同时利用可逆加成-断裂链转聚合的方式合成了聚甲基丙烯酸二甲基氨基乙酯-聚甲基丙烯酸酯线型共聚物(pdma-b-pma),接下来利用脱硫-点击反应将pdma-b-pma与csq-th相连得到含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]并赋予该聚合物双亲的特性。接下来利用一种改进的界面合成方案,在fe
3+
催化下从端噻吩基团开始生长聚3,4-乙撑二氧噻吩(edot)链段,形成倍半硅氧烷基星型可溶性共轭聚合物(csq-(pdma-b-pma)-pedot)。
[0075]
其中,相关反应物、中间产物和终产物的结构式如下:
[0076]
八乙烯基笼型倍半硅氧烷(csq),其结构式为:
[0077][0078]
含多个端噻吩基团的八面体倍半硅氧烷(csq-th),其结构式为:
[0079][0080]
含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th],其结构式为:
[0081]
[0082]
其中,50≤m≤100;1≤n1≤10;
[0083]
含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot],其结构式为:
[0084][0085]
其中,50≤m≤100;1≤n1≤10;5≤n2≤20。
[0086]
在本发明的一些实例中,星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成方法包括如下步骤:
[0087]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0088]
dl-n-乙酰高半胱氨酸硫内酯和4-二甲氨基吡啶溶于二氧六环中,移入schlenk烧瓶中,通氮气保护。加入2-噻吩乙胺,将反应液进行三次冷冻脱气循环,置于油浴反应器中反应。接下来将已经脱气后的csq/二氧六环溶液注入反应瓶,继续在油浴反应器中反应。反应结束后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,层析柱分离,真空干燥,获得目标产物。
[0089]
其中,dl-n-乙酰高半胱氨酸硫内酯和二氧六环按质量体积比为0.1~0.3g:1ml进行配比;4-二甲氨基吡啶和二氧六环按质量体积比为0.003~0.005g:1ml进行配比;2-噻吩乙胺和二氧六环按质量体积比为0.03~0.05g:1ml进行配比。
[0090]
csq/二氧六环溶液浓度为0.1~0.5mmol/ml,优选0.2~0.3mmol/ml;csq/二氧六环溶液由csq和二氧六环按体积比为0.05~1:1,优选0.2~0.8:1进行配比。
[0091]
步骤(1)中,dl-n-乙酰高半胱氨酸硫内酯与2-噻吩乙胺的反应时间、以及加入csq/二氧六环溶液后的反应时间独立为8~20小时,该两步反应的温度独立为30~50℃。真空干燥温度为25~30℃,真空干燥时间约24小时。
[0092]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0093]
甲基丙烯酸n,n-二甲基氨基乙酯(dma)、s-十二烷基-s
’‑
(r,r
’‑
二甲基-r
”‑
乙酸)三硫代碳酸盐(dttc)和偶氮二异丁腈(aibn)溶于二氧六环,移入schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。油浴锅中反应。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥后获得目标产物。
[0094]
其中,s-十二烷基-s
’‑
(r,r-二甲基-r
”‑
乙酸)三硫代碳酸盐和二氧六环按质量体积比为0.03~0.05g:1ml进行配比;甲基丙烯酸n,n-二甲基氨基乙酯和二氧六环按质量体积比为1.2~1.8g:1ml进行配比;偶氮二异丁晴和二氧六环按质量体积比为0.0014~0.0018g:1ml进行配比;石油醚和二氧六环按体积比为10~20:1进行配比。
[0095]
步骤(2)中的所述搅拌速度300~600r/min,反应时间为2~6小时,反应温度为65~75℃。真空干燥温度为25~30℃,真空干燥时间约24小时。s-十二烷基-s
’‑
(r,r
’‑
二甲基-r
”‑
乙酸)三硫代碳酸盐(dttc)的合成方法可参考macromolecules(2002),volume 35.issue 18,pages 6754-6756。
[0096]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0097]
聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma),丙烯酸甲酯(ma)和偶氮二异丁腈(aibn)溶于二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中。反应结束后用液氮猝灭反应。解冻后将反应液缓慢滴加入冰石油醚中沉淀,快速搅拌,静置后取沉淀。真空干燥后获得目标产物。
[0098]
其中,聚甲基丙烯酸n,n-二甲基氨基乙酯和二氧六环按质量体积比为0.2~0.4g:1ml进行配比;丙烯酸甲酯和二氧六环按质量体积比为0.05~0.08g:1ml进行配比;偶氮二异丁晴和二氧六环按质量体积比为0.0004~0.0008g:1ml进行配比;石油醚和二氧六环按体积比为10~20:1进行配比。
[0099]
步骤(3)中的搅拌速度300~600r/min,反应时间为2~6小时,反应温度为65~75℃。真空干燥温度为25~30℃,干燥时间约24小时。
[0100]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]的合成:
[0101]
pdma-b-pma溶于二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦和丙胺,再进行3次冷冻脱气循环。油浴加热反应。接下来减压抽去溶剂和未反应的丙胺,注入脱气后的二氧六环,溶解后再减压抽去溶剂。接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液,然后反应器置于油浴中反应。随后减压抽去溶剂,将粘稠物溶于二氯甲烷中,缓慢滴入石油醚。将沉淀物真空干燥后得到目标产物。
[0102]
其中,pdma-b-pma和二氧六环按质量体积比为0.1~0.3g:1ml进行配比;三丁基膦和二氧六环按体积为0.5~1.5:1进行配比;丙胺和二氧六环按体积比为0.2~0.6:1进行配比。csq-th/三乙胺/二氧六环溶液和溶解pdma-b-pma的二氧六环按体积比为0.8~1.2:1进行配比。csq-th/三乙胺/二氧六环溶液中,csq-th和二氧六环按质量体积比为0.002~0.006g:1ml进行配比,三乙胺和二氧六环按体积比为0.01~0.02:1进行配比。
[0103]
步骤(4)中的搅拌速度300~600r/min,pdma-b-pma加入三丁基膦和丙胺后的反应时间为3~5小时;加入csq-th/三乙胺/二氧六环溶液后的反应时间为12~36小时。步骤(4)
的两步反应的温度为30~50℃。真空干燥温度为25~30℃,干燥时间约24小时。
[0104]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0105]
csq-(pdma-b-pma)-th溶于盐酸溶液,加入三氯化铁溶液。将混合液移入血清瓶中,尽量不要碰到内壁。然后将3,4-乙撑二氧噻吩(edot)/正丁醇溶液沿瓶内壁缓慢覆盖在水相上。在室温下避光静置反应后,取出下层蓝色水相,高速离心,取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中,快速搅拌。当没有气泡溢出时,静置。移取上层棕色清液,浓缩后在冰石油醚中沉淀,取出沉淀,真空干燥得到目标产物。
[0106]
其中,csq-(pdma-b-pma)-th和正丁醇按质量体积比为0.008~0.012g:1ml进行配比;盐酸水溶液和正丁醇按体积比为0.3~0.4:1进行配比;3,4-乙撑二氧噻吩和正丁醇按质量体积比为0.002~0.003g:1进行配比;nahco3/thf溶液和正丁醇按体积比为1.2~1.6:1进行配比;石油醚和正丁醇按体积比为10~20:1进行配比。
[0107]
步骤(5)中反应时间为24~36小时,反应温度为20~30℃。所述的盐酸水溶液为0.5~1.5mol/l。所述的三氯化铁水溶液的浓度为0.1~0.2mol/l。所述的nahco3/thf液为0.5~1.5mol/l nahco3水溶液与四氢呋喃混合液,nahco3水溶液和四氢呋喃按体积比为1:2~4进行配比。真空干燥温度为25~30℃,干燥时间约24小时。
[0108]
以下结合具体的实施例进一步说明本发明的技术方案。以下实施例中所用的原料,如无特殊说明,均可从常规商业途径得到;所采用的工艺,如无特殊说明,均采用本领域的常规工艺。本发明所述室温或者常温,如无特殊说明,均指25
±
5℃。
[0109]
实施例1
[0110]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0111]
dl-n-乙酰高半胱氨酸硫内酯(1.908g,12mmol)和4-二甲氨基吡啶(0.109g,0.9mmol)溶于18ml二氧六环中,移入50ml schlenk烧瓶中,通氮气30分钟。加入2-噻吩乙胺(1.143g,9mmol),将反应液进行三次冷冻脱气循环,置于40℃油浴反应器中反应8小时。
[0112]
接下来将已经脱气后的csq/二氧六环溶液(5ml,0.24mmol/ml)注入反应瓶,继续在40℃油浴反应器中反应14小时。然后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入20ml二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,再层析柱分离,常温真空干燥24小时,获得淡黄色粘稠状液体。提纯后产物产率为62%。
[0113]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0114]
dma(15.7g,0.1mol)、dttc(0.364g,1mmol)和aibn(0.0164g,0.1mmol)溶于10ml二氧六环,移入50ml schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。65℃油浴锅中反应2小时。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0115]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0116]
pdma(8.5g,0.6mmol),丙烯酸甲酯(2.58g,0.03mol)和aibn(0.0164g,0.1mmol)溶于36ml二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中,反应温度为65℃,持续搅拌2小时。随后用液氮猝灭反应。解冻后将反应液缓慢滴加入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0117]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]
的合成:
[0118]
pdma-b-pma(7.5g,0.5mmol)溶于36ml二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦(1.04ml,5mmol)和丙胺(0.411ml,5mmol),再进行3次冷冻脱气循环。油浴加热反应,温度为40℃,持续搅拌4小时。接下来减压抽去溶剂和未反应的丙胺,注入20ml脱气后的二氧六环,溶解后再减压抽去溶剂。
[0119]
接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液(1.310g/0.505ml/30ml),然后反应器置于油浴中,反应温度为30℃,持续搅拌12小时。随后减压抽去溶剂,将粘稠物溶于10ml二氯甲烷中,缓慢滴入180ml石油醚沉淀溶真空干燥后得到淡黄色固体。提纯后产物产率为95%。
[0120]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0121]
csq-(pdma-b-pma)-th(0.1g,1.3
×
10-3
mmol)溶于3.5ml盐酸溶液(1mol/l),加入1.5ml三氯化铁溶液(0.1mol/l),再加入0.5ml四氢呋喃。将混合液移入30ml血清瓶中,尽量不要碰到内壁。然后将10ml edot/正丁醇溶液(0.02mol/l)沿瓶内壁缓慢覆盖在水相上。
[0122]
在室温下避光静置反应24小时后,取出下层蓝色水相,高速离心5分钟(12400rad/min),取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中(15ml 1mol/l/45ml thf),快速搅拌。当没有气泡溢出时,继续搅拌0.5小时,然后静置2小时。移取上层棕色清液,浓缩后在冰石油醚中沉淀,沉淀真空干燥48小时,得到褐色固体。提纯后产物产率为95%。
[0123]
其中csq-th和csq-(pdma-b-pma)-pedot的1h-nmr图谱分别如图1和2所示,csq-(pdma-b-pma)-pedot的红外图谱如图3所示。1h-nmr图谱和红外图谱反映本实施例成功合成了结构为预想结构的的csq-th,以及csq-(pdma-b-pma)-pedot。利用尺寸排除色谱测试得到csq-(pdma-b-pma)-pedot的质均分子量为36843da,mw/mn=1.81。
[0124]
同时,以铂丝为对电极,玻碳电极为工作电极,甘汞电极为参比电极。将csq-(pdma-b-pma)-pedot涂覆在玻碳电极中,以0.1mol/l kcl水溶液为电解液测试,得到csq-(pdma-b-pma)-pedot的循环伏安曲线图如图4所示,图4循环曲线有明显的氧化还原峰且能够形成闭合的回路,能够证明csq-(pdma-b-pma)-pedot具有电化学活性(具有一定的导电性),说明通过使用不含柔性链段的噻吩单体可获得可溶的、具有导电性质的材料。基于所具有的导电性能,csq-(pdma-b-pma)-pedot有望应用于有机光电材料制备技术领域。
[0125]
另外,经试验,csq-(pdma-b-pma)-pedot可溶于大部分有机溶剂,例如四氢呋喃、乙醇、丙酮等,其中csq-(pdma-b-pma)-pedot溶解在四氢呋喃中的照片如图5所示。
[0126]
实施例2
[0127]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0128]
dl-n-乙酰高半胱氨酸硫内酯(1.908g,12mmol)和4-二甲氨基吡啶(0.109g,0.9mmol)溶于18ml二氧六环中,移入50ml schlenk烧瓶中,通氮气30分钟。加入2-噻吩乙胺(1.143g,9mmol),将反应液进行三次冷冻脱气循环,置于40℃油浴反应器中反应10小时。
[0129]
接下来将已经脱气后的csq/二氧六环溶液(5ml,0.24mmol/ml)注入反应瓶,继续在40℃油浴反应器中反应16小时。然后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入20ml二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,
再层析柱分离,常温真空干燥24小时,获得淡黄色粘稠状液体。提纯后产物产率为64%。
[0130]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0131]
dma(15.7g,0.1mol)、dttc(0.364g,1mmol)和aibn(0.0164g,0.1mmol)溶于10ml二氧六环,移入50ml schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。65℃油浴锅中反应3小时。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0132]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0133]
pdma(9.5g,0.6mmol),丙烯酸甲酯(2.58g,0.03mol)和aibn(0.0164g,0.1mmol)溶于36ml二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中,反应温度为65℃,持续搅拌2小时。随后用液氮猝灭反应。解冻后将反应液缓慢滴加入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0134]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]的合成:
[0135]
pdma-b-pma(8.0g,0.5mmol)溶于36ml二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦(1.04ml,5mmol)和丙胺(0.411ml,5mmol),再进行3次冷冻脱气循环。油浴加热反应,温度为40℃,持续搅拌4小时。接下来减压抽去溶剂和未反应的丙胺,注入20ml脱气后的二氧六环,溶解后再减压抽去溶剂。
[0136]
接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液(1.310g/0.505ml/30ml),然后反应器置于油浴中,反应温度为30℃,持续搅拌14小时。随后减压抽去溶剂,将粘稠物溶于10ml二氯甲烷中,缓慢滴入180ml石油醚沉淀溶真空干燥后得到淡黄色固体。提纯后产物产率为92%。
[0137]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0138]
csq-(pdma-b-pma)-th(0.1g,1.2
×
10-3
mmol)溶于3.5ml盐酸溶液(1mol/l),加入1.5ml三氯化铁溶液(0.1mol/l),再加入0.5ml四氢呋喃。将混合液移入30ml血清瓶中,尽量不要碰到内壁。然后将10ml edot/正丁醇溶液(0.02mol/l)沿瓶内壁缓慢覆盖在水相上。
[0139]
在室温下避光静置反应24小时后,取出下层蓝色水相,高速离心5分钟(12400rad/min),取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中(15ml 1mol/l/45ml thf),快速搅拌。当没有气泡溢出时,继续搅拌0.5小时,然后静置2小时。移取上层棕色清液,浓缩后在冰石油醚中沉淀,沉淀真空干燥48小时,得到褐色固体。提纯后产物产率为92%。
[0140]
实施例2的csq-(pdma-b-pma)-pedot的循环伏安曲线也有明显的氧化还原峰且能够形成闭合的回路,也具有一定的导电性,同时在不同有机溶剂中也有良好的溶解性。利用尺寸排除色谱测试得到csq-(pdma-b-pma)-pedot的质均分子量为26442da,mw/mn=1.66。
[0141]
实施例3
[0142]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0143]
dl-n-乙酰高半胱氨酸硫内酯(1.908g,12mmol)和4-二甲氨基吡啶(0.109g,0.9mmol)溶于18ml二氧六环中,移入50ml schlenk烧瓶中,通氮气30分钟。加入2-噻吩乙胺(1.143,9mmol),将反应液进行三次冷冻脱气循环,置于40℃油浴反应器中反应12小时。
[0144]
接下来将已经脱气后的csq/二氧六环溶液(5ml,0.24mmol/ml)注入反应瓶,继续在40℃油浴反应器中反应14小时。然后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入20ml二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,再层析柱分离,常温真空干燥24小时,获得淡黄色粘稠状液体。提纯后产物产率为65%。
[0145]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0146]
dma(15.7g,0.1mol)、dttc(0.364g,1mmol)和aibn(0.0164g,0.1mmol)溶于10ml二氧六环,移入50ml schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。65℃油浴锅中反应4小时。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0147]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0148]
pdma(10g,0.6mmol),丙烯酸甲酯(2.58g,0.03mol)和aibn(0.0164g,0.1mmol)溶于36ml二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中,反应温度为65℃,持续搅拌2小时。随后用液氮猝灭反应。解冻后将反应液缓慢滴加入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0149]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]的合成:
[0150]
pdma-b-pma(8g,0.5mmol)溶于36ml二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦(1.04ml,5mmol)和丙胺(0.411ml,5mmol),再进行3次冷冻脱气循环。油浴加热反应,温度为40℃,持续搅拌4小时。接下来减压抽去溶剂和未反应的丙胺,注入20ml脱气后的二氧六环,溶解后再减压抽去溶剂。
[0151]
接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液(1.310g/0.505ml/30ml),然后反应器置于油浴中,反应温度为30℃,持续搅拌12小时。随后减压抽去溶剂,将粘稠物溶于10ml二氯甲烷中,缓慢滴入180ml石油醚沉淀溶真空干燥后得到淡黄色固体。提纯后产物产率为93%。
[0152]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0153]
csq-(pdma-b-pma)-th(0.1g,1.2
×
10-3
mmol)溶于3.5ml盐酸溶液(1mol/l),加入1.5ml三氯化铁溶液(0.1mol/l),再加入0.5ml四氢呋喃。将混合液移入30ml血清瓶中,尽量不要碰到内壁。然后将10ml edot/正丁醇溶液(0.02mol/l)沿瓶内壁缓慢覆盖在水相上。
[0154]
在室温下避光静置反应24小时后,取出下层蓝色水相,高速离心5分钟(12400rad/min),取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中(15ml 1mol/l/45ml thf),快速搅拌。当没有气泡溢出时,继续搅拌0.5小时,然后静置2小时。移取上层棕色清液,浓缩后在冰石油醚中沉淀,沉淀真空干燥48小时,得到褐色固体。提纯后产物产率为92%。
[0155]
实施例3的csq-(pdma-b-pma)-pedot的循环伏安曲线也有明显的氧化还原峰且能够形成闭合的回路,也具有一定的导电性,同时在不同有机溶剂中也有良好的溶解性。利用尺寸排除色谱测试得到csq-(pdma-b-pma)-pedot的质均分子量为40361da,mw/mn=1.86。
[0156]
实施例4
[0157]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0158]
dl-n-乙酰高半胱氨酸硫内酯(1.908g,12mmol)和4-二甲氨基吡啶(0.109g,0.9mmol)溶于18ml二氧六环中,移入50ml schlenk烧瓶中,通氮气30分钟。加入2-噻吩乙胺(1.143g,9mmol),将反应液进行三次冷冻脱气循环,置于40℃油浴反应器中反应10小时。
[0159]
接下来将已经脱气后的csq/二氧六环溶液(5ml,0.24mmol/ml)注入反应瓶,继续在40℃油浴反应器中反应10小时。然后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入20ml二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,再层析柱分离,常温真空干燥24小时,获得淡黄色粘稠状液体。提纯后产物产率为61%。
[0160]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0161]
dma(15.7g,0.1mol)、dttc(0.364g,1mmol)和aibn(0.0164g,0.1mmol)溶于10ml二氧六环,移入50ml schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。65℃油浴锅中反应2.5小时。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0162]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0163]
pdma(9.0g,0.6mmol),丙烯酸甲酯(2.58g,0.03mol)和aibn(0.0164g,0.1mmol)溶于36ml二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中,反应温度为65℃,持续搅拌3小时。随后用液氮猝灭反应。解冻后将反应液缓慢滴加入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0164]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]的合成:
[0165]
pdma-b-pma(7.8g,0.5mmol)溶于36ml二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦(1.04ml,5mmol)和丙胺(0.411ml,5mmol),再进行3次冷冻脱气循环。油浴加热反应,温度为40℃,持续搅拌4小时。接下来减压抽去溶剂和未反应的丙胺,注入20ml脱气后的二氧六环,溶解后再减压抽去溶剂。
[0166]
接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液(1.310g/0.505ml/30ml),然后反应器置于油浴中,反应温度为30℃,持续搅拌12小时。随后减压抽去溶剂,将粘稠物溶于10ml二氯甲烷中,缓慢滴入180ml石油醚沉淀溶真空干燥后得到淡黄色固体。提纯后产物产率为92%。
[0167]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0168]
csq-(pdma-b-pma)-th(0.1g,1.2
×
10-3
mmol)溶于3.5ml盐酸溶液(1mol/l),加入1.5ml三氯化铁溶液(0.1mol/l),再加入0.5ml四氢呋喃。将混合液移入30ml血清瓶中,尽量不要碰到内壁。然后将10ml edot/正丁醇溶液(0.02mol/l)沿瓶内壁缓慢覆盖在水相上。
[0169]
在室温下避光静置反应24小时后,取出下层蓝色水相,高速离心5分钟(12400rad/min),取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中(15ml 1mol/l/45ml thf),快速搅拌。当没有气泡溢出时,继续搅拌0.5小时,然后静置2小时。移取上层棕色清液,浓缩后在冰石油醚中沉淀,沉淀真空干燥48小时,得到褐色固体。提纯后产物产率为95%。
[0170]
实施例4的csq-(pdma-b-pma)-pedot的循环伏安曲线也有明显的氧化还原峰且能够形成闭合的回路,也具有一定的导电性,同时在不同有机溶剂中也有良好的溶解性。利用
尺寸排除色谱测试得到csq-(pdma-b-pma)-pedot的质均分子量为33721da,mw/mn=1.75。
[0171]
实施例5
[0172]
(1)含多个端噻吩基团的八面体倍半硅氧烷(csq-th)的合成:
[0173]
dl-n-乙酰高半胱氨酸硫内酯(1.908g,12mmol)和4-二甲氨基吡啶(0.109g,0.9mmol)溶于18ml二氧六环中,移入50ml schlenk烧瓶中,通氮气30分钟。加入2-噻吩乙胺(1.143g,9mmol),将反应液进行三次冷冻脱气循环,置于40℃油浴反应器中反应12小时。
[0174]
接下来将已经脱气后的csq/二氧六环溶液(5ml,0.24mmol/ml)注入反应瓶,继续在40℃油浴反应器中反应12小时。然后减压抽走溶剂,用大量去离子水洗涤粘稠物3次。弃去水溶液,再加入20ml二氯甲烷溶解粘稠物,加入4a分子筛干燥溶液。过滤取滤液,浓缩后,再层析柱分离,常温真空干燥24小时,获得淡黄色粘稠状液体。提纯后产物产率为60%。
[0175]
(2)聚甲基丙烯酸n,n-二甲基氨基乙酯(pdma)的合成:
[0176]
dma(15.7g,0.1mol)、dttc(0.364g,1mmol)和aibn(0.0164g,0.1mmol)溶于10ml二氧六环,移入50ml schlenk反应瓶,快速搅拌均匀后,进行3次冷冻脱气循环。70℃油浴锅中反应3小时。用液氮猝灭反应,将冷冻的反应瓶置于温水中融化,反应液滴入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0177]
(3)聚甲基丙烯酸n,n-二甲基氨基乙酯-聚丙烯酸甲酯(pdma-b-pma)的合成:
[0178]
pdma(10.5g,0.6mmol),丙烯酸甲酯(2.58g,0.03mol)和aibn(0.0164g,0.1mmol)溶于36ml二氧六环,移入schlenk瓶。快速进行3次冷冻脱气循环后,将反应瓶置于油浴中,反应温度为65℃,持续搅拌2小时。随后用液氮猝灭反应。解冻后将反应液缓慢滴加入220ml冰石油醚中沉淀,快速搅拌,静置后倾去上层液体。沉淀物真空干燥48小时后获得黄色固体。
[0179]
(4)含多个端噻吩基团的倍半硅氧烷基星型杂化聚合物[csq-(pdma-b-pma)-th]的合成:
[0180]
pdma-b-pma(8.5g,0.5mmol)溶于36ml二氧六环,移入schlenk瓶,然后快速地进行3次冷冻脱气循环。再氩气保护下,注入三丁基膦(1.04ml,5mmol)和丙胺(0.411ml,5mmol),再进行3次冷冻脱气循环。油浴加热反应,温度为40℃,持续搅拌4小时。接下来减压抽去溶剂和未反应的丙胺,注入20ml脱气后的二氧六环,溶解后再减压抽去溶剂。
[0181]
接下来在氩气气氛保护下注入csq-th/三乙胺/二氧六环溶液(1.310g/0.505ml/30ml),然后反应器置于油浴中,反应温度为30℃,持续搅拌12小时。随后减压抽去溶剂,将粘稠物溶于10ml二氯甲烷中,缓慢滴入180ml石油醚沉淀溶真空干燥后得到淡黄色固体。提纯后产物产率为93%。
[0182]
(5)含多条共轭链段的倍半硅氧烷基星型可溶性共轭聚合物[csq-(pdma-b-pma)-pedot]的合成:
[0183]
csq-(pdma-b-pma)-th(0.1g,1.2
×
10-3
mmol)溶于3.5ml盐酸溶液(1mol/l),加入1.5ml三氯化铁溶液(0.1mol/l),再加入0.5ml四氢呋喃。将混合液移入30ml血清瓶中,尽量不要碰到内壁。然后将10ml edot/正丁醇溶液(0.02mol/l)沿瓶内壁缓慢覆盖在水相上。
[0184]
在室温下避光静置反应24小时后,取出下层蓝色水相,高速离心5分钟(12400rad/min),取上层蓝色清液。将清液滴入nahco3水溶液/四氢呋喃混合液中(15ml 1mol/l/45ml thf),快速搅拌。当没有气泡溢出时,继续搅拌0.5小时,然后静置2小时。移取上层棕色清
液,浓缩后在冰石油醚中沉淀,沉淀真空干燥48小时,得到褐色固体。提纯后产物产率为93%。
[0185]
实施例5的csq-(pdma-b-pma)-pedot的循环伏安曲线也有明显的氧化还原峰且能够形成闭合的回路,也具有一定的导电性,同时在不同有机溶剂中也有良好的溶解性。。利用尺寸排除色谱测试得到csq-(pdma-b-pma)-pedot的质均分子量为38490da,mw/mn=1.91。
[0186]
另外,比较发现,实施例1~5中的产物csq-(pdma-b-pma)-pedot的1h-nmr图谱化学位移在3.80~4.70ppm之间的质子氢数量不同,表明产物中edot数量的不同。且不同实施例中产物csq-(pdma-b-pma)-pedot的循环伏安曲线的氧化峰和还原峰位置有变化,表明产物的电化学性能不同,通过调控产物的分子量可调节其电化学性能。
[0187]
对比例1
[0188]
参考申请号为202010042371.1的中国专利,制得结构式如下的星型聚合物:
[0189]
其中50≤m≤100;1≤n1≤10;5≤n2≤20。
[0190]
经测试,该星型聚合物不具备导电性。通过与实施例1~5比较可见,本发明的星型可溶性共轭聚合物通过改变笼型倍半硅氧烷上的聚合链段,使聚合物表现出一定的导电性。
[0191]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1