一种两亲聚合物和脂溶性营养素纳米颗粒及其制备方法与流程
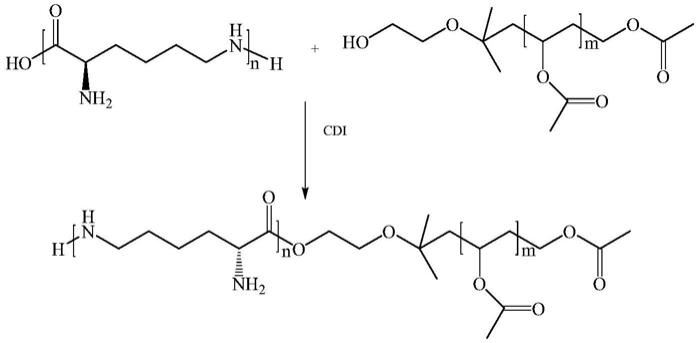
1.本发明属于营养素应用领域,具体涉及一种两亲聚合物和脂溶性营养素纳米颗粒及其制备方法。
背景技术:
2.脂溶性营养素包括辅酶q10、姜黄素、脂溶性维生素、类胡萝卜素等,其为维持机体繁殖、生长发育和生存等一切生命活动的必要物质。将脂溶性营养素采用纳米制剂技术制成纳米颗粒有利于提高其生物利用度。
3.然而,现有纳米制剂技术在实际运用中存在一些不可避免的痛点,例如,机械研磨粉碎(高压均质/气流粉碎)、喷雾干燥、热挤出制粒都需要较高的能量输入(生产过程的过热),而且这些工艺的粒径分布、聚团的倾向性、微粒表面的粗糙度、药物微粒里的非晶形区等不能较好地控制。近年来,随着人们对超临界流体性质认识的不断深入,超临界流体技术作为一种安全可靠、方便快速、可控制调节的微粒制备新技术而备受关注。
4.超临界溶液快速溶胀法(ress)是超临界流体制粒技术中的一个重要分支,适用于超临界co2流体互溶物质的微细化。由于二氧化碳是很弱的溶剂,绝大部分物质在超临界co2中溶解度较小,需要加入有机溶剂增强其溶解度。高分子量的两亲聚合物在超临界二氧化碳中溶解度极低,因此在使用ress制备纳米颗粒的过程中仍然不可避免需要使用有机溶剂增溶,这在一定程度上降低了超临界二氧化碳体系的优势。
技术实现要素:
5.本发明的目的之一在于克服现有技术采用超临界溶液快速溶胀法制备脂溶性营养素纳米颗粒过程中需要使用有机溶剂增溶从而不绿色环保的缺陷,而提供一种新的两亲聚合物,该两亲聚合物具有亲和超临界co2的特性,在采用超临界溶液快速溶胀法制备纳米颗粒过程中无需使用任何有机溶剂,能够充分发挥超临界流体制备纳米颗粒的优势,且所得脂溶性营养素纳米颗粒的包埋率较高,稳定性好,在体内的停留时间较长,生物利用度较高。
6.本发明的目的之二在于提供一种两亲聚合物的制备方法。
7.本发明的目的之三在于提供由上述方法制备得到的两亲聚合物。
8.本发明的目的之四在于提供一种含有上述两亲聚合物的脂溶性营养素纳米颗粒。
9.本发明的目的之五在于利用上述两亲聚合物的特性(亲水性、亲脂性、亲二氧化碳性)提供一种无需使用有机溶剂的超临界流体ress制粒技术制备的脂溶性营养素纳米颗粒的方法。
10.本发明的目的之六在于提供由上述方法制备得到的脂溶性营养素纳米颗粒。
11.具体地,本发明提供了一种两亲聚合物,其中,所述两亲聚合物包括聚赖氨酸链段与聚醋酸乙烯酯链段,所述聚赖氨酸链段中赖氨酸的聚合度为21~35,所述聚醋酸乙烯酯链段中醋酸乙烯酯的聚合度为50~500。
12.在一种优选实施方式中,所述两亲聚合物的数均分子量为8000~50000。
13.本发明还提供了一种两亲聚合物的制备方法,其中,该方法包括以下步骤:
14.s11、将醋酸乙烯酯单体在端羟基链转移剂和自由基引发剂的存在下进行自由基聚合反应,得到聚合度为50~500且末端为羟基的聚醋酸乙烯酯;
15.s12、在无水及惰性气氛下,将聚合度为21~35的聚赖氨酸采用羰基二咪唑进行活化,得到活性聚赖氨酸中间体;
16.s13、将步骤s1所得聚醋酸乙烯酯与步骤s2所得活性聚赖氨酸中间体进行酯化反应,得到两亲聚合物。
17.在一种优选实施方式中,步骤s11中,所述自由基引发剂为偶氮类自由基引发剂。
18.在一种优选实施方式中,步骤s11中,所述端羟基链转移剂为异丙氧基乙醇。
19.在一种优选实施方式中,步骤s11中,所述醋酸乙烯酯单体、端羟基链转移剂和自由基引发剂的质量比为1:(5~15):(0.01~0.05)。
20.在一种优选实施方式中,步骤s11中,所述自由基聚合反应的条件包括反应温度为65~150℃,反应时间为8~15小时。
21.在一种优选实施方式中,步骤s12中,所述聚赖氨酸与羰基二咪唑的摩尔比为1:(1~1.35)。
22.在一种优选实施方式中,步骤s12中,所述活化的条件包括温度为25~35℃,时间为3~4小时。
23.在一种优选实施方式中,步骤s13中,所述聚醋酸乙烯酯与聚赖氨酸的质量比为(0.5~4):1。
24.在一种优选实施方式中,步骤s13中,所述酯化反应的条件包括反应温度为50~60℃,反应时间为4~18小时。
25.在一种优选实施方式中,步骤s13中,所述酯化反应的方式为将聚醋酸乙烯酯于50~80℃下溶于无水二甲基甲酰胺中,在惰性气氛及温度为5~15℃下缓慢滴加入步骤s2所得活性聚赖氨酸中间体中并搅拌反应5~20min,之后升温至50~60℃搅拌反应4~18小时,反应结束后将反应液倒入无水乙醇中得到白色沉淀,过滤后用冰的无水乙醇打浆洗涤,之后再冷冻干燥,得到两亲聚合物。
26.本发明还提供了由上述方法制备得到的两亲聚合物。
27.本发明还提供了一种脂溶性营养素纳米颗粒,其中,所述脂溶性营养素纳米颗粒中含有脂溶性营养素、上述两亲聚合物、聚氧乙烯氢化蓖麻油、维生素e聚乙二醇琥珀酸酯和益生元,所述脂溶性营养素纳米颗粒采用超临界溶液快速溶胀法制备得到。
28.在一种优选实施方式中,所述脂溶性营养素选自辅酶q10、姜黄素、脂溶性维生素和类胡萝卜素的至少一种。
29.在一种优选实施方式中,所述脂溶性维生素选自维生素a、维生素d、维生素e和维生素k中的至少一种。
30.在一种优选实施方式中,所述类胡萝卜素选自β-胡萝卜素、叶黄素、虾青素、番茄红素和玉米黄素中的至少一种。
31.在一种优选实施方式中,所述益生元选自低聚半乳糖、低聚果糖、异麦芽酮糖醇和异麦芽酮糖中的至少一种。
32.在一种优选实施方式中,所述脂溶性营养素与两亲聚合物的质量比为1:(1~30)。
33.在一种优选实施方式中,所述聚氧乙烯氢化蓖麻油与两亲聚合物的质量比为1:(5~10)。
34.在一种优选实施方式中,所述维生素e聚乙二醇琥珀酸酯与两亲聚合物的质量比为1:(0.5~2.5)。
35.在一种优选实施方式中,所述益生元与维生素e聚乙二醇琥珀酸酯的质量比为1:(0.1~1)。
36.本发明还提供了一种脂溶性营养素纳米颗粒的制备方法,该方法包括以下步骤:
37.s21、将脂溶性营养素、聚氧乙烯氢化蓖麻油以及上述两亲聚合物混合均匀,将所得混合溶液溶解于超临界二氧化碳中得到超临界溶液;
38.s22、将维生素e聚乙二醇琥珀酸酯和益生元溶于水中,得到混合水相溶液;
39.s23、将步骤s21所得超临界溶液喷射至步骤s22所得混合水相溶液中,经分散、沉淀和干燥,得到脂溶性营养素纳米颗粒。
40.在一种优选实施方式中,所述脂溶性营养素选自辅酶q10、姜黄素、脂溶性维生素和类胡萝卜素的至少一种。
41.在一种优选实施方式中,所述脂溶性维生素选自维生素a、维生素d、维生素e和维生素k中的至少一种。
42.在一种优选实施方式中,所述类胡萝卜素选自β-胡萝卜素、叶黄素、虾青素、番茄红素和玉米黄素中的至少一种。
43.在一种优选实施方式中,所述益生元选自低聚半乳糖、低聚果糖、异麦芽酮糖醇和异麦芽酮糖中的至少一种;
44.在一种优选实施方式中,所述脂溶性营养素与两亲聚合物的质量比为1:(1~30)。
45.在一种优选实施方式中,所述聚氧乙烯氢化蓖麻油与两亲聚合物的质量比为1:(5~10)。
46.在一种优选实施方式中,所述混合水相溶液中维生素e聚乙二醇琥珀酸酯和益生元的总浓度以质量分数计为5~15%。
47.在一种优选实施方式中,所述维生素e聚乙二醇琥珀酸酯与两亲聚合物的质量比为1:(0.5~2.5)。
48.在一种优选实施方式中,所述益生元与维生素e聚乙二醇琥珀酸酯的质量比为1:(0.1~1)。
49.在一种优选实施方式中,将所得混合溶液溶解于超临界二氧化碳的条件包括膨胀压力为15~35mpa,膨胀温度323~343k。
50.在一种优选实施方式中,所述喷射的速度为0.5-1.5ml/min。
51.本发明还提供了由上述方法制备得到的脂溶性营养素纳米颗粒。
52.本发明的有益效果:
53.(1)本发明在纳米颗粒制备过程无需使用任何有机溶剂,充分发挥了超临界流体ress制粒技术制备纳米颗粒的优势,绿色安全。
54.(2)本发明提供的两亲聚合物为阳离子聚合物,能够使得脂溶性纳米颗粒通过静电作用与体内带负电荷的生物大分子如蛋白质、脱氧核糖核酸、核糖核酸等结合,由此延长
脂溶性纳米颗粒在胃肠粘膜的停留时间。
55.(3)本发明提供的两亲聚合物由亲水链段和疏水链段两部分组成,由于疏水作用等非共价力的驱动使其在水中自组装时可将疏水物质如脂溶性营养素包载于内部,自组装形成具有“核-壳”结构的纳米颗粒,从而对脂溶性营养素起到较好的稳定作用。同时,益生元在维生素e聚乙二醇琥珀酸酯的乳化作用下吸附包埋在脂质体表面进行二次包埋,双层包埋可以更好地保护脂溶性营养素的活性,提高其生物利用度。因而,本发明提供的纳米颗粒包埋率高、不易聚集、稳定性好、生物利用度高。
56.(4)聚氧乙烯氢化蓖麻油具有短链的peg,可以提高纳米颗粒之间的排斥作用,防止其聚集而从一定程度上提高纳米颗粒的稳定性。然而,短链peg容易被吞噬细胞摄取而失去作用,而两亲性的高分子聚合物因其特殊的长链结构可以有效延长体内长循环时间,促进纳米颗粒在血液中的循环,不被吞噬系统识别而失去作用,因此,将聚氧乙烯氢化蓖麻油和具有本发明特定结构的两亲聚合物复配能够有效提高纳米颗粒的稳定性及体内长循环时间。
57.(5)本发明提供的两亲聚合物制备过程以羰基二咪唑作为缩合剂,反应结束后只生成咪唑小分子和二氧化碳,副产物很容易洗脱除去。
具体实施方式
58.本发明提供的两亲聚合物包括聚赖氨酸链段与聚醋酸乙烯酯链段。其中,所述聚赖氨酸链段的具体结构如式(1)所示,其为亲水链段,由21~35个赖氨酸组成,也即,聚合度n为21~35,如21、22、23、24、25、26、27、28、29、30、31、32、33、34、35。所述聚醋酸乙烯酯链段的具体结构如式(2)所示,其为疏水链段,由50~500个醋酸乙烯酯组成,也即,聚合度m为50~500,如50、60、70、80、90、100、120、150、180、200、220、250、280、300、320、350、380、400、420、450、480、500。此外,所述两亲聚合物的数均分子量优选为8000~50000,如8000、10000、12000、15000、18000、20000、22000、25000、28000、30000、32000、35000、38000、40000、42000、45000、48000、50000。
[0059][0060]
ε-聚赖氨酸(ε-polylysine,ε-pl)因其良好的水溶性、广谱的抑菌性以及在体内可降解为易吸收的赖氨酸等特性而成为近年来较为热门且研究最为广泛的一种聚氨基酸,并已被广泛地应用于功能性食品及给药系统领域的研究中。在聚赖氨酸主链上引入聚醋酸乙烯酯链段,则可以弥补其分子链中亲脂性不足的缺陷,所得两亲聚合物既有氨基基团,又具有一定的亲脂能力,可在水溶液中自组装成为具有核-壳结构的纳米颗粒,能与药物发生亲和,对脂溶性营养素的纳米颗粒起到较好的稳定作用。此外,疏水链段选用与超临界二氧化碳亲和的聚醋酸乙烯酯链段,能够赋予两亲聚合物亲和超临界二氧化碳性能,从而为后续采用超临界流体ress制粒技术制备脂溶性营养素纳米颗粒的过程中无需使用任何有机溶剂奠定了基础,能够充分发挥超临界流体ress制粒技术的优势,绿色安全。此外,由特定聚合度的聚赖氨酸链段与聚醋酸乙烯酯链段形成的两亲聚合物因其特殊的长链结构可以
有效延长体内循环时间,弥补聚氧乙烯氢化蓖麻油由于短链peg容易被吞噬细胞摄取而失去作用的缺陷,从而有效提高纳米颗粒的稳定性及体内长循环时间。
[0061]
本发明提供的两亲聚合物的制备方法包括以下步骤:s11、将醋酸乙烯酯单体(vac)在端羟基链转移剂和自由基引发剂的存在下进行自由基聚合反应,得到聚合度为50~500且末端为羟基的聚醋酸乙烯酯(pvac-oh);s12、在无水及惰性气氛下,将聚合度为21~35的聚赖氨酸采用羰基二咪唑进行活化,得到活性聚赖氨酸中间体;s13、将步骤s1所得聚醋酸乙烯酯与步骤s2所得活性聚赖氨酸中间体进行酯化反应,得到两亲聚合物(ε-pl-pvac)。
[0062]
在本发明中,步骤s11所采用的自由基引发剂可以为现有的各种能够引发醋酸乙烯酯单体实现自由基聚合反应的引发剂,例如,可以选自偶氮类引发剂、过氧化物类引发剂和氧化还原类引发剂中的至少一种,优选为偶氮类引发剂。所述偶氮类引发剂的具体实例包括但不限于:偶氮二异丁酸二甲酯、偶氮二异丁脒盐酸盐、偶氮二甲酰胺、偶氮二异丙基咪唑啉盐酸盐、偶氮异丁氰基甲酰胺、偶氮二环己基甲腈(accn)、偶氮二氰基戊酸、偶氮二异丙基咪唑啉、偶氮二异丁腈(aibn)、偶氮二异戊腈和偶氮二异庚腈中的至少一种。此外,本发明对自由基引发剂的用量没有特别地限制,但为了兼顾引发速率与聚合产物分子量的大小,所述自由基引发剂与醋酸乙烯酯单体的质量比优选为(0.01~0.05):1。
[0063]
在本发明中,步骤s11所采用的端羟基链转移剂可以为现有的各种能够使链增长自由基发生转移的含端羟基的化合物,例如,优选为异丙醇(ipa)和/或异丙氧基乙醇(ipe),特别优选为异丙氧基乙醇。此外,所述端羟基链转移剂与醋酸乙烯酯单体的质量比优选为(5~15):1。以所述端羟基链转移剂为异丙氧基乙醇(ipe)为例,所述自由基聚合反应过程如下所示:
[0064][0065]
本发明对步骤s11中自由基聚合反应的条件没有特别的限定,只有能够使得醋酸乙烯酯单体实现自由基聚合以获得聚醋酸乙烯酯链段即可,通常包括反应温度可以为65~150℃,反应时间可以为8~15小时。此外,所述自由基聚合反应通常在惰性气体保护下进行,目的是为了克服氧阻聚,使得聚合反应顺利进行。所述惰性气体是指不与反应物和产物发生化学反应的任意一种气体或气体混合物,如氮气、氦气和元素周期表零族气体中的至少一种。
[0066]
在本发明中,步骤s12中采用羰基二咪唑(cdi)对聚赖氨酸中的羧基进行活化,得到活性聚赖氨酸中间体。其中,所述聚赖氨酸与羰基二咪唑的摩尔比优选为1:(1~1.35)。所述活化的条件通常包括温度可以为25~35℃,时间可以为3~4小时。以cdi作为缩合剂对聚赖氨酸进行活化,反应结束后只生成咪唑小分子和二氧化碳,副产物很容易洗脱除去。此外,所述活化在无水及惰性气氛下进行。其中,保持无水的方式通常可以包括采用加热方式对反应容器进行干燥以及采用冷冻干燥、分子筛除水除氧、手套箱抽换气等方式对反应原料进行除水。保持惰性气氛的方式通常为往反应容器中通入惰性气体或者抽真空。
[0067]
在本发明中,步骤s13中将聚醋酸乙烯酯与活性聚赖氨酸中间体进行酯化反应。其中,所述聚醋酸乙烯酯与聚赖氨酸的质量比优选为(0.5~4):1。所述酯化反应的条件通常包括反应温度可以为50~60℃,反应时间可以为4~18小时。在一种具体实施方式中,所述酯化反应的方式为将聚醋酸乙烯酯于50~80℃下溶于无水二甲基甲酰胺中,在惰性气氛及温度为5~15℃下缓慢滴加入活性聚赖氨酸中间体中并搅拌反应5~20min,之后升温至50~60℃搅拌反应4~18小时,反应结束后将反应液倒入无水乙醇中得到白色沉淀,过滤后用冰的无水乙醇打浆洗涤,之后再冷冻干燥,得到两亲聚合物,具体反应过程如下所示:
[0068][0069]
本发明还提供了由上述方法制备得到的两亲聚合物。
[0070]
本发明提供的脂溶性营养素纳米颗粒中含有脂溶性营养素、上述两亲聚合物、聚氧乙烯氢化蓖麻油、维生素e聚乙二醇琥珀酸酯和益生元。其中,所述脂溶性营养素与两亲聚合物的质量比优选为1:(1~30),如1:1、1:3、1:5、1:8、1:10、1:12、1:15、1:18、1:20、1:22、1:25、1:28、1:30等。所述聚氧乙烯氢化蓖麻油与两亲聚合物的质量比优选为1:(5~10),如1:5、1:6、1:7、1:8、1:9、1:10等。所述维生素e聚乙二醇琥珀酸酯与两亲聚合物的质量比优选为1:(0.5~2.5),如1:0.5、1:0.8、1:1、1:1.2、1:1.5、1:1.8、1:2.0、1:2.2、1:2.5等。所述益生元与维生素e聚乙二醇琥珀酸酯的质量比优选为1:(0.1~1),如1:0.1、1:0.2、1:0.3、1:0.4、1:0.5、1:0.6、1:0.7、1:0.8、1:0.9、1:1.0等。所述脂溶性营养素纳米颗粒采用超临界溶液快速溶胀法(ress)制得。所述脂溶性营养素纳米颗粒的包封率优选为90%以上,更优选为90~95%。所述脂溶性营养素纳米颗粒的数均粒径优选为80~100nm,粒径分布范围优选为65~115nm。
[0071]
所述脂溶性营养素可以为现有的各种不溶于水而溶于脂肪及非极性有机溶剂的从外界环境中摄取的可给人体提供能量、机体构成成分和组织修复以及生理调节功能的化学成分,其具体实例包括但不限于:辅酶q10、姜黄素、脂溶性维生素和类胡萝卜素的至少一种。其中,所述脂溶性维生素可以选自维生素a、维生素d、维生素e和维生素k中的至少一种。所述类胡萝卜素可以选自β-胡萝卜素、叶黄素、虾青素、番茄红素和玉米黄素中的至少一种。
[0072]
所述聚氧乙烯氢化蓖麻油中聚氧乙烯链段的分子量优选为200以下,更优选为100以下。所述聚氧乙烯氢化蓖麻油的具体实例包括但不限于:peg-10氢化蓖麻油、peg-20氢化蓖麻油、peg-40氢化蓖麻油、peg-45氢化蓖麻油、peg-50氢化蓖麻油和peg-100氢化蓖麻油中的至少一种,特别优选为peg-40氢化蓖麻油。所述聚氧乙烯氢化蓖麻油中所具有的短链
peg能够提高纳米颗粒之间的排斥作用,防止纳米颗粒之间的聚集,提高其稳定性。
[0073]
所述维生素e聚乙二醇琥珀酸酯(tpgs)为维生素e的水溶性衍生物,由维生素e琥珀酸酯的羧基与聚乙二醇的羟基反应而成,其中既含有维生素e亲酯基团,又含有聚乙二醇亲水长链,因而具有较好的表面活性剂性质及水溶性,不仅能够起乳化作用以吸附包埋脂质体,而且还能够增加脂溶性营养素在胃肠道中的吸收,提高生物利用度。所述维生素e聚乙二醇琥珀酸酯的具体实例包括但不限于:聚乙二醇200维生素e琥珀酸酯、聚乙二醇400维生素e琥珀酸酯、聚乙二醇600维生素e琥珀酸酯、聚乙二醇800维生素e琥珀酸酯、聚乙二醇1000维生素e琥珀酸酯、聚乙二醇2000维生素e琥珀酸酯中的至少一种。
[0074]
所述益生元是一种不能被人体消化酶消化却能刺激某种益生菌的数量和活性从而改善人体健康的有机物质,其具体实例包括但不限于:低聚半乳糖、低聚果糖、异麦芽酮糖醇和异麦芽酮糖中的至少一种。
[0075]
本发明提供的脂溶性营养素纳米颗粒的制备方法包括以下步骤:s21、将脂溶性营养素、聚氧乙烯氢化蓖麻油以及上述两亲聚合物混合均匀,将所得混合溶液溶解于超临界二氧化碳中得到超临界溶液;s22、将维生素e聚乙二醇琥珀酸酯和益生元溶于水中,得到混合水相溶液;s23、将步骤s21所得超临界溶液喷射至步骤s22所得混合水相溶液中,经分散、沉淀和干燥,得到脂溶性营养素纳米颗粒。其中,所述脂溶性营养素与两亲聚合物的质量比优选为1:(1~30),如1:1、1:3、1:5、1:8、1:10、1:12、1:15、1:18、1:20、1:22、1:25、1:28、1:30等。所述聚氧乙烯氢化蓖麻油与两亲聚合物的质量比优选为1:(5~10),如1:5、1:6、1:7、1:8、1:9、1:10等。所述混合水相溶液中维生素e聚乙二醇琥珀酸酯和益生元的总浓度以质量分数计优选为5~15%,如5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%等。所述维生素e聚乙二醇琥珀酸酯与两亲聚合物的质量比优选为1:(0.5~2.5),如1:0.5、1:0.8、1:1、1:1.2、1:1.5、1:1.8、1:2.0、1:2.2、1:2.5等。所述益生元与维生素e聚乙二醇琥珀酸酯的质量比优选为1:(0.1~1),如1:0.1、1:0.2、1:0.3、1:0.4、1:0.5、1:0.6、1:0.7、1:0.8、1:0.9、1:1.0等。此外,所述脂溶性营养素、聚氧乙烯氢化蓖麻油、维生素e聚乙二醇琥珀酸酯和益生元的种类已经在上文中有所描述,在此不作赘述。
[0076]
本发明对将所得混合溶液溶解于超临界二氧化碳的条件没有特别的限定,通常包括膨胀压力优选为15~35mpa,膨胀温度优选为323~343k。在本发明中,所述压力均指表压。此外,所述喷射的速度优选为0.5-1.5ml/min。
[0077]
在一种具体实施方式中,所述脂溶性营养素纳米颗粒的制备方法包括以下步骤:s21、将脂溶性营养素、聚氧乙烯氢化蓖麻油以及上述两亲聚合物超声混合,之后将所得混合溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体,之后由高压泵加压后进入超临界反应釜中,将压力控制在15~35mpa并将温度控制在323~343k,混合溶液溶解于超临界二氧化碳中得到超临界溶液;s22、将维生素e聚乙二醇琥珀酸酯和益生元溶于水中,得到混合水相溶液;s23、待体系达到平衡后,快速释放压力,超临界溶液通过喷嘴(直径为400~600μm,温度为70~90℃)以0.5-1.5ml/min的速度喷射至混合水相溶液中,经分散、沉淀和干燥,得到脂溶性营养素纳米颗粒。
[0078]
本发明还提供了由上述方法制备得到的脂溶性营养素纳米颗粒。
[0079]
以下将通过实施例对本发明进行详细描述。
[0080]
以下实施例和对比例中所采用的检测仪器包括:ms3000马尔文激光粒度分析仪,
购自英国马尔文;高效液相色谱仪,购自安捷伦;bt-1000粉体综合特性测试仪,购自丹东百特仪器有限公司。
[0081]
以下实施例和对比例中,粒径和包封率按照以下方法测定:
[0082]
(1)粒径采用ms3000马尔文激光粒度分析仪检测,具体地:称取1g样品于100ml烧杯中,冲入50ml温水(30℃)并充分搅拌,观察溶解过程,复乳液的颜色及其表面是否有结膜或浮油现象,最后将复乳液采用ms3000马尔文激光粒度分析仪测定油滴粒径大小。
[0083]
(2)包封率(%)通过产品有效成分含量以及表面有效成分含量进行计算,其中,表面有效成分含量是指未被包埋的那部分有效成分,使用反透析法分离出表面游离有效成分,包封率/%=(产品有效成分含量-表面有效成分含量)/产品有效成分含量
×
100%。
[0084]
制备例1制备两亲聚合物
[0085]
s11、含羟基端的聚醋酸乙烯酯pvac-oh制备:将50.0g醋酸乙烯酯单体(vac)和2.5g偶氮二异丁腈(aibn)加入到400ml异丙氧基乙醇(ipe)中,再将所得反应溶液加热至65℃搅拌12小时,反应结束后减压除去溶剂,加入200ml冷的正己烷沉淀,过滤得到固体,之后将得到的固体溶解在50ml丙酮中,并再次加入200ml冷正己烷中再次沉淀。重复两次,然后在真空下干燥得到46.5g粘性固体产物聚醋酸乙烯酯pvac-oh,聚合度为150。
[0086]
s12、在无水及惰性气氛下,用无水二甲基甲酰胺溶解预干燥的46.5g聚赖氨酸(南京凯轩生物科技有限公司公司生产,数均分子量为4100,聚合度为28)配制成浓度为0.8g/ml的溶液,然后加入cdi(聚赖氨酸与cdi的摩尔比为1:1.35),在25℃下搅拌反应4小时,得到活性聚赖氨酸中间体溶液。
[0087]
s13、将46.5g聚醋酸乙烯酯pvac-oh于55℃下溶于无水二甲基甲酰胺中,在惰性气氛中,在低温5℃下缓慢滴加入活性聚赖氨酸中间体溶液中,滴加完毕后搅拌10min,然后升温至60℃搅拌反应10小时;通过薄层色谱跟踪反应进程,反应结束后将反应液倒入无水乙醇中得到白色沉淀。反应液与无水乙醇体积比为1:8;过滤后用冰的无水乙醇打浆洗涤3次,最后冷冻干燥得到两亲聚合物ε-pl-pvac,记为pl-pvac-1,其数均分子量为18000,产量为42.13g,收率为45.3%。
[0088]1h nmr(400mhz,cdcl3):δ5.07-4.66,4.38-4.17,4.15-4.02,3.69-3.44,2.87-2.74,2.71-2.61,2.57-2.44,2.26-1.95,1.92-1.50,1.48-1.38,1.35-1.24,1.21-1.06,0.97-0.81.
[0089]
从1h nmr的结果结合反应机理可以看出,由该制备例所得两亲聚合物pl-pvac-1包括聚赖氨酸链段与聚醋酸乙烯酯链段。
[0090]
制备例2制备两亲聚合物
[0091]
s11、含羟基端的聚醋酸乙烯酯pvac-oh制备:将25.0g醋酸乙烯酯单体(vac)和0.65g偶氮二环己基甲腈(accn)加入到250ml异丙氧基乙醇(ipe)中,再将所得反应溶液加热至70℃搅拌9小时,反应结束后减压除去溶剂,加入200ml冷的正己烷沉淀,过滤得到固体,之后将得到的固体溶解在45ml丙酮中,并再次加入175ml冷正己烷中再沉淀。重复两次,然后在真空下干燥得到23.4g粘性固体的产物聚醋酸乙烯酯pvac-oh,聚合度为60。
[0092]
s12、在无水及惰性气氛下,用无水二甲基甲酰胺溶解预干燥的39g聚赖氨酸(南京凯轩生物科技有限公司公司生产,数均分子量为4100,聚合度为28)配制成浓度为0.6g/ml的溶液,然后加入cdi(聚赖氨酸与cdi的摩尔比为1:1.2),在35℃下搅拌反应3小时,得到活
性聚赖氨酸中间体溶液。
[0093]
s13、将23.4g聚醋酸乙烯酯pvac-oh于55℃下溶于无水二甲基甲酰胺中,在惰性气氛中,在低温10℃下缓慢滴加入活性聚赖氨酸中间体溶液中,滴加完毕后搅拌10min,然后升温至55℃搅拌反应12小时;通过薄层色谱跟踪反应进程,反应结束后将反应液倒入无水乙醇中得到白色沉淀,反应液与无水乙醇体积比为1:12;过滤后用冰的无水乙醇打浆洗涤3次,最后冷冻干燥得到两亲聚合物ε-pl-pvac,记为pl-pvac-2,其数均分子量为10000,产量为27.2g,收率为43.6%。
[0094]1h nmr(400mhz,cdcl3):δ5.06-4.79,4.75-4.54,4.41-4.01,3.67-3.46,3.43-3.30,3.27-3.01,2.32-1.93,1.76-1.48,1.44-1.37,1.34-1.24,1.21-1.10,1.04-0.79.
[0095]
从1h nmr的结果结合反应机理可以看出,由该制备例所得两亲聚合物pl-pvac-2包括聚赖氨酸链段与聚醋酸乙烯酯链段。
[0096]
制备例3制备两亲聚合物
[0097]
s11、含羟基端的聚醋酸乙烯酯pvac-oh制备:将64.0g醋酸乙烯酯单体(vac)和0.64g偶氮二异丁腈(aibn)加入到650ml异丙氧基乙醇(ipe)中,再将所得反应溶液加热至85℃搅拌14小时,反应结束后减压除去溶剂,加入220ml冷的正己烷沉淀,过滤得到固体,之后将得到的固体溶解在60ml丙酮中,并再次加入220ml冷正己烷中再沉淀。重复两次,然后在真空下干燥得到62.5g粘性液体产物聚醋酸乙烯酯pvac-oh,聚合度为400。
[0098]
s12、在无水及惰性气氛下,用无水二甲基甲酰胺溶解预干燥的25g聚赖氨酸(南京凯轩生物科技有限公司公司生产公司生产,数均分子量为4700,聚合度为32)配制成浓度为0.4g/ml的溶液,然后加入cdi(聚赖氨酸与cdi的摩尔比为1:1.0),在35℃下搅拌反应4小时,得到活性聚赖氨酸中间体溶液。
[0099]
s13、将62.5g聚醋酸乙烯酯pvac-oh于55℃下溶于无水二甲基甲酰胺中,在惰性气氛中,在低温5℃下缓慢滴加入活性聚赖氨酸中间体溶液中,滴加完毕后搅拌10min,然后升温至60℃搅拌反应12小时;通过薄层色谱跟踪反应进程,反应结束后将反应液倒入无水乙醇中得到白色沉淀,反应液与无水乙醇体积比为1:15;过滤后用冰的无水乙醇打浆洗涤3次,最后冷冻干燥得到两亲聚合物ε-pl-pvac,记为pl-pvac-3,其数均分子量在41000,产量为41.4g,收率为47.3%。
[0100]1h nmr(400mhz,cdcl3):δ5.07-4.59,4.56-4.15,4.11-4.01,3.53-3.36,2.80-2.69,2.63-2.51,2.48-2.36,2.14-1.86,1.62-1.33,1.26-1.09,0.94-0.77.
[0101]
从1h nmr的结果结合反应机理可以看出,由该制备例所得两亲聚合物pl-pvac-3包括聚赖氨酸链段与聚醋酸乙烯酯链段。
[0102]
对比制备例1制备两亲聚合物
[0103]
在氮气氛围下,将29gε-多聚赖氨酸(ε-pl)(南京凯轩生物科技有限公司公司生产,数均分子量为4100,聚合度为25~31)、36g生育酚琥珀酸酯(tos)、100ml无水二甲基甲酰胺(dmf)以及465ul催化剂n-n二异丙基碳二甲酰亚胺(dic)于45℃下搅拌反应20h,待反应完成后,经过层析分离、透析提纯、减压旋蒸、冷冻干燥,得到参比两亲聚合物,记为d-εpl-tos复合物。
[0104]
实施例1采用超临界ress法制备辅酶q10纳米颗粒
[0105]
s21、将7g两亲聚合物pl-pvac-1、0.87g聚氧乙烯氢化蓖麻油rh-40及0.35g辅酶
q10超声混合,之后将所得辅酶q10溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在30mpa且将温度控制在333k,混合溶液溶解于超临界二氧化碳中得到辅酶q10超临界溶液。
[0106]
s22、将5g维生素e聚乙二醇1000琥珀酸酯(tpgs)和5g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0107]
s23、待体系达到平衡后,快速释放压力,辅酶q10超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为89nm、粒径分布范围为80nm~110nm、包封率约为91.8%的辅酶q10纳米颗粒,记为w1。
[0108]
实施例2采用超临界ress法制备辅酶q10纳米颗粒
[0109]
s21、将4g两亲聚合物pl-pvac-2、0.8g聚氧乙烯氢化蓖麻油rh-40及4g辅酶q10超声混合,之后将所得辅酶q10溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在35mpa且将温度控制在323k,混合溶液溶解于超临界二氧化碳中得到辅酶q10超临界溶液。
[0110]
s22、将2g维生素e聚乙二醇1000琥珀酸酯(tpgs)和8g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0111]
s23、待体系达到平衡后,快速释放压力,辅酶q10超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为96nm、粒径分布范围为81nm~115nm、包封率约为91.3%的辅酶q10纳米颗粒,记为w2。
[0112]
实施例3采用超临界ress法制备辅酶q10纳米颗粒
[0113]
s21、将5g两亲聚合物pl-pvac-3、0.55g聚氧乙烯氢化蓖麻油rh-40及0.5g辅酶q10超声混合,之后将所得辅酶q10溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在35mpa且将温度控制在338k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到辅酶q10超临界溶液。
[0114]
s22、将2g维生素e聚乙二醇1000琥珀酸酯(tpgs)和10g异麦芽酮糖溶于水中,配成水相溶液100ml。
[0115]
s23、待体系达到平衡后,快速释放压力,辅酶q10超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1.5ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为86nm、粒径分布范围为75nm~107nm、包封率约为92.5%的辅酶q10纳米颗粒,记为w3。
[0116]
实施例4采用超临界ress法制备姜黄素纳米颗粒
[0117]
s21、将4g两亲聚合物pl-pvac-1、0.4g peg-20氢化蓖麻油及0.4g姜黄素超声混合,之后将所得姜黄素溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在25mpa且将温度控制在338k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到姜黄素超临界溶液。
[0118]
s22、将5g维生素e聚乙二醇800琥珀酸酯(tpgs)和5g异麦芽酮糖醇溶于水中,配成
混合水相溶液100ml。
[0119]
s23、待体系达到平衡后,快速释放压力,姜黄素超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为88.6nm、粒径分布范围为74nm~103nm、包封率约为92.7%的姜黄素纳米颗粒,记为w4。
[0120]
实施例5采用超临界ress法制备β-胡萝卜素纳米颗粒
[0121]
s21、将5g两亲聚合物pl-pvac-2、0.65g聚氧乙烯氢化蓖麻油rh-40及0.35gβ-胡萝卜素超声混合,之后将所得β-胡萝卜素溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在33mpa且将温度控制在333k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到β-胡萝卜素超临界溶液。
[0122]
s22、将3g维生素e聚乙二醇800琥珀酸酯(tpgs)和6g低聚果糖溶于水中,配成混合水相溶液100ml。
[0123]
s23、待体系达到平衡后,快速释放压力,β-胡萝卜素超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为82nm、粒径分布范围为73nm~105nm、包封率约为92.4%的β-胡萝卜素纳米颗粒,记为w5。
[0124]
实施例6采用超临界ress法制备维生素a纳米颗粒
[0125]
s21、将3g两亲聚合物pl-pvac-3、0.6g peg-45氢化蓖麻油及0.1g维生素a超声混合,之后将所得维生素a溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在15mpa且将温度控制在323k,同时开启磁力搅拌器(25kw),混合溶液溶解于超临界二氧化碳中得到维生素a超临界溶液。
[0126]
s22、将2g维生素e聚乙二醇600琥珀酸酯(tpgs)和3g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0127]
s23、待体系达到平衡后,快速释放压力,维生素a超临界溶液通过喷嘴(直径为500μm,温度为80℃)以0.5ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为91.5nm、粒径分布范围为83nm~109nm、包封率约为91.3%的维生素a纳米颗粒,记为w6。
[0128]
实施例7采用超临界ress法制备玉米黄素纳米颗粒
[0129]
s21、将5g两亲聚合物pl-pvac-1、1g聚氧乙烯氢化蓖麻油rh-40及0.2g玉米黄素超声混合,之后将所得玉米黄素溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在35mpa且将温度控制在338k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到玉米黄素超临界溶液。
[0130]
s22、将2g维生素e聚乙二醇600琥珀酸酯(tpgs)和4g异麦芽酮糖溶于水中,配成混合水相溶液100ml。
[0131]
s23、待体系达到平衡后,快速释放压力,玉米黄素超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得
到数均粒径为96nm、粒径分布范围为81nm~115nm、包封率约为91.3%的玉米黄素纳米颗粒,记为w7。
[0132]
实施例8采用超临界ress法制备叶黄素纳米颗粒
[0133]
s21、将4g两亲聚合物pl-pvac-1、0.65g聚氧乙烯氢化蓖麻油rh-40及0.27g叶黄素超声混合,之后将所得叶黄素溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入反应釜中,将压力控制在33mpa且将温度控制在333k,同时开启磁力搅拌器(35kw),混合溶液溶解于超临界二氧化碳中得到叶黄素超临界溶液。
[0134]
s22、将5g维生素e聚乙二醇1000琥珀酸酯(tpgs)和10g低聚果糖溶于水中,配成混合水相溶液100ml。
[0135]
s23、待体系达到平衡后,快速释放压力,叶黄素超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为84nm、粒径分布范围为72nm~101nm、包封率约为93.1%的叶黄素纳米颗粒,记为w8。
[0136]
实施例9采用超临界ress法制备番茄红素纳米颗粒
[0137]
s21、将3g两亲聚合物pl-pvac-1、0.4g peg-50氢化蓖麻油及2g番茄红素超声混合,之后将所得番茄红素溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入反应釜中,将压力控制在33mpa且将温度控制在338k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到番茄红素超临界溶液。
[0138]
s22、将5g维生素e聚乙二醇800琥珀酸酯(tpgs)和5g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0139]
s23、待体系达到平衡后,快速释放压力,番茄红素超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为89nm、粒径分布范围为69nm~105nm、包封率约为92.2%的番茄红素纳米颗粒,记为w9。
[0140]
实施例10采用超临界ress法制备维生素d3纳米颗粒
[0141]
s21、将3g两亲聚合物pl-pvac-1、0.5g聚氧乙烯氢化蓖麻油rh-40及0.4g维生素d3超声混合,之后将所得维生素d3溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入反应釜中,将压力控制在25mpa且将温度控制在343k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到维生素d3超临界溶液。
[0142]
s22、将3g维生素e聚乙二醇400琥珀酸酯(tpgs)和3g异麦芽酮糖醇溶于水中,配成混合水相溶液100ml。
[0143]
s23、待体系达到平衡后,快速释放压力维生素d3超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为88nm、粒径分布范围为74nm~107nm、包封率约为92.6%的维生素d3纳米颗粒,记为w10。
[0144]
实施例11采用超临界ress法制备维生素k2纳米颗粒
[0145]
s21、将2g两亲聚合物pl-pvac-1、0.25g聚氧乙烯氢化蓖麻油rh-40及2g维生素k2超声混合,之后将所得维生素k2溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在35mpa且将温度控制在338k,同时开启磁力搅拌器(30kw),混合溶液溶解于超临界二氧化碳中得到维生素k2超临界溶液。
[0146]
s22、将4g维生素e聚乙二醇1000琥珀酸酯(tpgs)和4g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0147]
s23、待体系达到平衡后,快速释放压力,维生素k2超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为96nm、粒径分布范围为84nm~110nm、包封率约为90.7%的维生素k2纳米颗粒,记为w11。
[0148]
实施例12采用超临界ress法制备维生素e纳米颗粒
[0149]
s21、将3g两亲聚合物pl-pvac-1、0.5g聚氧乙烯氢化蓖麻油rh-40及2g维生素e超声混合,之后将所得维生素e溶液送入超临界反应釜中;开启二氧化碳钢瓶,二氧化碳气体经制冷机冷却成液体后,由高压泵加压后进入超临界反应釜中,将压力控制在30mpa且将温度控制在333k,同时开启磁力搅拌器(35kw),混合溶液溶解于超临界二氧化碳中得到维生素e超临界溶液。
[0150]
s22、将2g维生素e聚乙二醇1000琥珀酸酯(tpgs)和8g低聚半乳糖溶于水中,配成混合水相溶液100ml。
[0151]
s23、待体系达到平衡后,快速释放压力,维生素e超临界溶液通过喷嘴(直径为500μm,温度为80℃)以1ml/min的速度喷射至混合水相溶液中,经分散、沉淀、干燥,即可得到数均粒径为93nm、粒径分布范围为81nm~107nm、包封率约为91.7%的维生素e纳米颗粒,记为w12。
[0152]
对比例1采用超临界ress法制备辅酶q10纳米颗粒
[0153]
按照实施例1的方法制备辅酶q10纳米颗粒,不同的是,将两亲聚合物pl-pvac-1采用相同重量份的聚赖氨酸替代,其余条件同实施例1,得到参比辅酶q10纳米颗粒,记为dw1,其数均粒径为287nm,粒径分布范围为135nm~417nm,包封率约为37%。
[0154]
对比例2采用超临界ress法制备辅酶q10纳米颗粒
[0155]
按照实施例1的方法制备辅酶q10纳米颗粒,不同的是,将维生素e聚乙二醇1000琥珀酸酯(tpgs)采用相同重量份的单硬脂酸甘油酯替代,其余条件同实施例1,得到参比辅酶q10纳米颗粒,记为dw2,其数均粒径为102nm,粒径分布范围为89~135nm,包封率约为87%。
[0156]
对比例3采用超临界ress法制备辅酶q10纳米颗粒
[0157]
按照实施例1的方法制备辅酶q10纳米颗粒,不同的是,将聚氧乙烯氢化蓖麻油rh-40采用单硬脂酸甘油酯替代,其余条件同实施例1,得到参比辅酶q10纳米颗粒,记为dw3,其数均粒径为119nm,粒径分布范围为92~148nm,包封率约为82%。
[0158]
对比例4采用超临界ress法制备辅酶q10纳米颗粒
[0159]
按照实施例1的方法制备辅酶q10纳米颗粒,不同的是,将两亲聚合物pl-pvac-1采用相同重量份的由对比制备例1所得参比两亲聚合物d-εpl-tos替代,其余条件同实施例1,得到参比辅酶q10纳米颗粒,记为dw4,其数均粒径为265nm,粒径分布范围为122nm~389nm,
包封率约为41%。
[0160]
对比例5采用喷雾干燥法制备辅酶q10纳米颗粒
[0161]
按照实施例1中的各组分配方制备辅酶q10纳米粉,不同的是,采用普通喷雾干燥法制备,具体步骤如下:将两亲聚合物pl-pvac-1、聚氧乙烯氢化蓖麻油rh-40及辅酶q10一起在研磨机中研磨至结晶颗粒至5微米以下,得到辅酶q10分散液;将维生素e聚乙二醇1000琥珀酸酯(tpgs)和低聚半乳糖溶于水中,配成混合水相溶液,加热至45℃并保温。然后混合,高速剪切乳化至油滴粒径≤2微米后,使用压力为60mpa的高压均质机均质至油滴粒径小于100纳米的乳液;最后在进风温度180℃,出风温度90℃条件下进行喷雾干燥得到参比辅酶q10纳米粉,记为dw5,其数均粒径为113nm,粒径分布范围为94nm~187nm,包封率约为85%。
[0162]
测试例1:在超临界二氧化碳中溶解性试验
[0163]
称取一定量由制备例1~3所得两亲聚合物、对比制备例1所得两亲聚合物以及聚赖氨酸分别装入高压平衡釜,然后将高压平衡釜接入管路并放入恒温水浴中,打开二氧化碳钢瓶,利用高压泵将高压钢瓶中的二氧化碳打入高压缓冲瓶,随后进入预热釜中,在其中被加热至实验设定温度(30℃)。经过预热的二氧化碳从底部不断流入离压平衡釜中,直到离压平衡釜中的压力达到实验所需压力(20mpa)。当温度与压力均达到实验要求后开始稳压计时,当平衡釜中的体系达到相平衡60分钟后,打开取样减压阀进行取样,溶有溶质的超临界二氧化碳从高压平衡釜顶端流出,流入双u型管,超临界二氧化碳在其中变为气体,而溶解在其中的溶质便在u型管中析出,二氧化碳气体则先后流经转子流量计和湿式气体流量计,经后者计量体积后排空,实验完成后,卸下u型管,测定u型管中溶质的质量。溶解度=(w
0-w1)/w0×
100%,其中,w0为最初加入高压平衡釜中的样品干重,w1为最终残留在u型管中的溶质干重。所得结果见表1。
[0164]
表1
[0165]
聚合物30℃、20mpa条件下溶于超临界二氧化碳的量聚赖氨酸小于0.2wt%pl-pvac-19.6wt%pl-pvac-27.5wt%pl-pvac-38.9wt%d-εpl-tos1.5wt%
[0166]
测试例2:稳定性试验
[0167]
分别将以上各实施例和对比例所得纳米颗粒装入密封无透光的小瓶中,置于25℃(恒温箱)条件下做加速老化实验,1份作为对照,一份放置10天,一份放置20天,一份放置30天,检测芯材脂溶性营养素含量,考察各纳米颗粒的稳定性能。结果如表2所示。
[0168]
表2
[0169][0170]
表2结果显示,超临界ress技术制备的脂溶性营养素纳米颗粒能够显著提高芯材的稳定性,且以本发明提供的两亲聚合物所得纳米颗粒的稳定性能最好。
[0171]
测试例3:体内药代动力学实验
[0172]
本发明提供的辅酶q10纳米颗粒经动物性实验证实,可以明显提高辅酶q10在体内的循环时间和生物利用度,具体实验步骤和结果如下:
[0173]
(1)样品,总共6个:
[0174]
样品样:实施例1制备的辅酶q10纳米颗粒;
[0175]
参比样1:对比例1制备的辅酶q10纳米颗粒;
[0176]
参比样2:对比例2制备的辅酶q10纳米颗粒;
[0177]
参比样3:对比例3制备的辅酶q10纳米颗粒;
[0178]
参比样4:对比例4制备的辅酶q10纳米颗粒;
[0179]
参比样5:对比例5制备的辅酶q10纳米颗粒。
[0180]
(2)实验动物与分组:
[0181]
由长沙市开福区东创实验动物科技服务部(实验动物使用许可证号为syxk(湘)2010-0010)提供的spf级昆明种雄性小鼠,鼠龄3个月,体重18-22g。按照国际实验动物实验准则对动物进行操作,以减少实验动物在实验过程中的痛苦。采用完全随机设计将大鼠随
机每10只分为一组,共6组。
[0182]
(3)实验条件:
[0183]
采用屏蔽环境进行动物实验,实验期间环境温度为23℃-24℃,湿度为50%-56%,每日自由摄取去离子水和标准饲料。
[0184]
(4)口服给药及样品采集:
[0185]
取禁食12h的小鼠随机10只分为一组,共6组。分别将样品样、参比样1、参比样2、参比样3、参比样4和参比样5按30mg/kg剂量给予小鼠灌胃处理,每组每个时间点(给药后5min、15min、30min、1h、2h、4h、6h、12h)分别取血于肝素抗凝管中,离心分离处理后进行测样。
[0186]
(5)药代动力学实验:
[0187]
小鼠分别经口服(30mg/kg)给予样品样、参比样1、参比样2、参比样3、参比样4和参比样5后,对实验所得的血浆样品处理后进样hplc分析测定。根据血药浓度结果进行拟合分析,计算血药浓度数据。所得结果见表3和表4。
[0188]
表3:小鼠经口服给药样品平均血药浓度-时间(mean
±
sd,n=5)(mg/l)
[0189]
采样时间样品样参比样1参比样2参比样3参比样4参比样55min10.72
±
1.263.22
±
1.347.42
±
1.266.23
±
1.174.08
±
0.475.08
±
0.5215min14.32
±
2.016.45
±
1.6511.98
±
2.0710.55
±
2.017.93
±
0.318.13
±
0.3830min20.22
±
2.1410.67
±
2.0118.58
±
2.0517.99
±
1.5511.55
±
2.1412.56
±
2.251h23.97
±
1.7912.55
±
1.2121.67
±
0.9620.43
±
1.1313.67
±
2.5314.11
±
2.472h16.03
±
3.338.12
±
1.1514.54
±
1.2513.92
±
1.039.75
±
1.4710.75
±
1.374h10.22
±
2.525.31
±
1.718.39
±
1.037.57
±
1.655.98
±
0.795.98
±
0.996h5.87
±
1.661.37
±
1.213.93
±
1.693.11
±
1.772.35
±
0.342.85
±
0.4112h3.31
±
1.330.89
±
0.312.15
±
1.521.79
±
2.111.05
±
0.291.45
±
0.32
[0190]
表4:小鼠经口服给药样品药动力学参数
[0191][0192]
血药浓度-时间曲线下面积(auc)是评定生物利用度的最可靠的指标。由表4可知样品样的auc比参比样的比值大。样品样与参比样1、2、4相比,超临界辅酶q10纳米颗粒的auc(o-t)和auc(0-∞)分别提高了100.3%/100.5%、12.9%/14.1%和68.0%/69.8%,表明本发明提供的两亲聚合物ε-pl-pvac可以更好地保护脂溶性物质的活性,且具备在体内长时间循环的特性,提高生物利用度。样品样与参比样3相比,超临界辅酶q10纳米颗粒的auc(o-t)和auc(0-∞)分别提高了21.6%和24.8%,表明本发明采用两亲聚合物与短链的聚氧乙烯氢化蓖麻油rh-40复配能使最终所得纳米颗粒具备更好的稳定性,从而提高生物利用度。样品样与参比样5相比,超临界辅酶q10纳米颗粒的auc(o-t)和auc(0-∞)分别提高了38.2%%和39.5%,表明超临界流体制粒技术相对于常规制粒技术制备出的纳米颗粒,具有更小的粒径使其在肠胃中粘附接触和快速被吸收,提高体内暴露程度,从而提高其吸收性和生物利用度。
[0193]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例
性的,不能理解为对本发明的限制,本领域的普通技术人员在不脱离本发明的原理和宗旨的情况下在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1