一种可回收生物基聚酯的制备方法
1.本发明涉及高分子材料及化学化工领域,具体的,本发明涉及一种聚(δ-己内酯)的制备方法。
背景技术:
2.在过去的近百年,由于各种塑料制品的飞速发展,我们的生活在许多方面都得到了极大的改善。然而如今大多数的塑料都具有线性的生命周期:它们来自石油,使用后直接废弃,没有得到有效的处理和回收,造成了资源的浪费和严重的环境污染。常用的废弃塑料处理方式为焚烧和填埋,少部分废弃塑料通过物理方法进行回收,但多次机械加工常常导致塑料性能的堕化。
3.解决高分子材料回收利用问题的一种手段为发展化学可回收高分子材料。化学可回收高分子材料指在较温和的反应条件及较低的能耗条件下可完全解聚得到其单体的高分子材料。目前对于这类高分子材料的研究仍然较少,文献报道的这类材料仅有聚(γ-丁内酯)、聚(反式六氢苯并呋喃-1(3h)酮)、聚(α-亚甲基-γ-丁内酯)、4-羟基脯氨酸衍生的聚硫内酯几种(nat.chem.2016,8,42-49;science 2018,360,398-403;j.am.chem.soc.2016,138,14326-14337;j.am.chem.soc.2019,141,4928-4935)。
4.δ-己内酯是一种来源广泛、价格低廉的生物基单体,可以从生物质原料中得到,是一种可再生原材料。目前报道的关于δ-己内酯进行开环聚合的催化剂包括有机酸、强碱、有机金属催化剂等,但它们存在着催化活性低、聚合反应时间长、催化剂毒性大、所得聚酯分子量低且不可控等问题(macromolecular chemistry and physics 2002,203,889
–
899;polymer chemistry 2015,6,2659
–
2660;macromolecules 2016,49,2419
–
2428;european polymer journal 2020,134,109858)。
5.有鉴于此,本发明提供了一种新型二元催化体系,实现δ-己内酯活性可控开环聚合,制备高分子量聚(δ-己内酯)的新方法。本发明提供的方法与以往报道的方法相比,具有以下有益之处:1)所使用的催化剂生物毒性低,并且易于从产物中除去,实验证明所得产物无明显细胞毒性,能够用于生物医药领域;2)所用催化体系活性高,能够实现δ-己内酯的快速可控开环聚合,制备高分子量聚(δ-己内酯);3)聚合反应不需要溶剂,符合绿色化学的原则;4)所得到的聚(δ-己内酯)可以定量解聚回收得到δ-己内酯单体。
技术实现要素:
6.本发明的目的是提供一种δ-己内酯快速可控开环聚合,制备高分子量聚(δ-己内酯)的方法,包括如下步骤:
7.将引发剂、强碱、二元脲和δ-己内酯混合均匀,在一定温度下反应一段时间,加入酸性物质终止反应,减压蒸馏除去未反应的δ-己内酯得到聚(δ-己内酯)。
8.上述制备方法中,所述聚(δ-己内酯)的化学结构式如式(ⅰ)所示:
[0009][0010]
其特征在于,n为大于等于5的自然数,r1为羟基、烷氧基或芳基烷氧基,具体可为羟基、甲氧基、乙氧基、异丙氧基、叔丁氧基、苄氧基、苯基乙氧基、苯基丙氧基、二苯基甲氧基、2,2-二苯基乙氧基。
[0011]
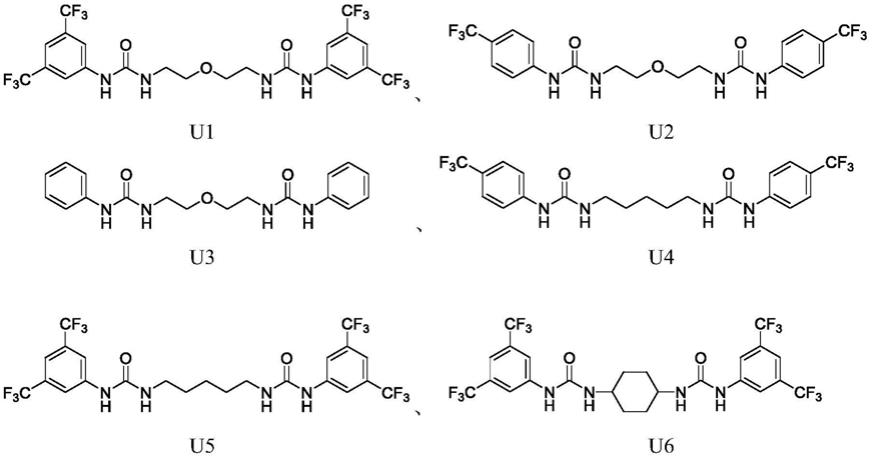
上述的制备方法中,二元脲有下列之一的结构:
[0012][0013]
上述制备方法中,所述引发剂为甲醇、乙醇、异丙醇、叔丁醇、苄醇、苯乙醇、苯丙醇、二苯基甲醇、2,2-二苯基乙醇、乙二醇、1,4-苯基二甲醇、丙三醇、季戊四醇;所述强碱为碱金属、碱金属化合物或有机磷腈碱催化剂,具体可为钠、钾、氢化钾、氢化钠、六[三(二甲基胺)磷氮烯]三聚磷腈({[(nme2)3p=n]2p=n}3)、磷腈配体p4-叔丁基([(nme2)3p=n]3p=ntbu,tert-bu-p4)、磷腈配体p2-叔丁基([(nme2)3p=n](nme2)2p=ntbu,tert-bu-p2);所述强碱与引发剂的摩尔比例为1/3~20/1;所述强碱与二元脲的摩尔比例为1/0.5~1/10。
[0014]
上述制备方法中,反应温度为10~50℃;所述反应时间为1~60min,所述引发剂与δ-己内酯的摩尔比例为1/10~1/3000。
[0015]
上述制备方法中,所述酸性物质为乙酸、苯甲酸、盐酸、硫酸、磷酸,所述酸性物质与强碱的摩尔比例为1/1~10/1。
附图说明
[0016]
图1为实施例1中制得的聚(δ-己内酯)的1h nmr谱图。
[0017]
图2为实施例2中制得的聚(δ-己内酯)的1h nmr谱图。
[0018]
图3为实施例1中制得的聚(δ-己内酯)的
13
c nmr谱图。
[0019]
图4为实施例2中制得的聚(δ-己内酯)的
13
c nmr谱图。
[0020]
图5为对比实施例1和实施例1制得的聚(δ-己内酯)的gpc谱图。
[0021]
图6为实施例1至实施例3制得的聚(δ-己内酯)在扫描速率为10℃/min的dsc谱图。
[0022]
图7为实施例7中不同投料比制得的聚(δ-己内酯)的gpc谱图。
[0023]
图8为实施例8回收得到的δ-己内酯与原始单体的1h nmr叠加谱图。
具体实施方式
[0024]
下述实施案例对本发明进行具体描述,但本发明不限于这些实施案例。
[0025]
下述实施案例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0026]
对比实施例1
[0027]
将(0.05mmol,5.41mg)苄醇,(0.1mmol,36.75mg)磷腈配体p2-叔丁基、(0.3mmol,63.6mg)二苯基脲加入到反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行2h,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物。gpc测得数均分子量为19.5kg/mol,分子量分布为1.38,gpc谱图如图5所示。
[0028]
对比实施例2
[0029]
将(0.05mmol,5.41mg)苄醇,(0.1mmol,36.75mg)磷腈配体p2-叔丁基、(0.3mmol,85.8mg)1-环己基-3-(4-三氟甲基苯基)脲加入到反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行2h,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为20.6kg/mol,分子量分布为1.32。
[0030]
对比实施例3
[0031]
将(0.05mmol,5.41mg)苄醇,(0.1mmol,36.75mg)磷腈配体p2-叔丁基、(0.15mmol,53.18mg)1,1'-(oxybis(ethane-2,1-diyl))bis(3-cyclohexylurea)加入到反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行0.5h,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为21.2kg/mol,分子量分布为1.58。
[0032]
实施例1
[0033]
将(0.05mmol,5.41mg)苄醇,(0.1mmol,36.75mg)磷腈配体p2-叔丁基、(0.15mmol,92.1mg)u1加入到反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行20min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯),其核磁氢谱如图1所示,核磁碳谱如图3所示。gpc测得数均分子量为100.09kg/mol,分子量分布为1.09,gpc谱图如图5所示,dsc谱图如图6所示。与单脲的催化体系相比(对比实施例1和对比实施例2),具有合适结构的二元脲的催化体系活性更高,反应时间更短,分子量提高到10万且分布很窄。
[0034]
实施例2
[0035]
将(0.05mmol,3.51mg)甲醇钾、(0.15mmol,71.76mg)u2加入反应管中,然后用注射
器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行30min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯),其核磁氢谱如图2所示,核磁碳谱如图4所示。gpc测得数均分子量为110.2kg/mol,分子量分布为1.15。dsc谱图如图6所示。
[0036]
实施例3
[0037]
(0.05mmol,5.41mg)苄醇,(0.1mmol,118mg)六[三(二甲基胺)磷氮烯]三聚磷腈,(0.15mmol,51.36mg)u3加入反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行20min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为112.0kg/mol,分子量分布为1.18。dsc谱图如图6所示。
[0038]
实施例4
[0039]
(0.05mmol,5.41mg)苄醇,(0.1mmol,63.4mg)磷腈配体p4-叔丁基催化剂,(0.15mmol,71.46mg)u4加入反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行20min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为105.3kg/mol,分子量分布为1.21。
[0040]
实施例5
[0041]
将(0.05mmol,68.13mg)苯丙醇、(0.1mmol,36.75mg)磷腈配体p2-叔丁基、(0.15mmol,91.86mg)u5加入反应管中,然后用注射器将(50mmol,5.5ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行30min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为121.2kg/mol,分子量分布为1.17。
[0042]
实施例6
[0043]
将(0.05mmol,69mg)1,4-苯基二甲醇、(0.2mmol,8mg)氢化钾、(0.15mmol,93.66mg)u6加入反应管中加入反应管中,然后用注射器将(100mmol,11ml)δ-己内酯加入反应管中并进行搅拌。反应在氮气保护下进行30min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物为目标开环聚合产物,即聚(δ-己内酯)。gpc测得数均分子量为210.2kg/mol,分子量分布为1.19。
[0044]
实施例7
[0045]
为了证明反应的可控性,设计了不同投料比的实验。
[0046]
将(0.05mmol,5.41mg)苄醇,(0.05mmol,14.7mg)磷腈配体p2-叔丁基、(0.075mmol,46.08mg)u1加入到5个反应管中,然后用注射器将不同量的δ-己内酯(2.5mmol,5mmol,7.5mmol,10mmol,12.5mmol)分别加入5个反应管中并进行搅拌(即单体与引发剂的比例分别为:50/1,100/1,150/1,200/1,250/1)。反应在氮气保护下进行20min,加入10滴醋酸终止反应。减压蒸馏除去未反应的δ-己内酯得到聚合物,核磁表征聚合物均为目标开环聚合产物,即聚(δ-己内酯)。得到的5个聚合物样品的gpc测得的数据分别为:数均分子量为6.2kg/mol,分子量分布为1.09;数均分子量为11.4kg/mol,分子量分布为1.15;数均分子量为16.0kg/mol,分子量分布为1.13;数均分子量为20.3kg/mol,分子量分布为1.18;数均分子量为25.6kg/mol,分子量分布为1.10;gpc谱图如图7所示。
[0047]
实施例8
[0048]
将(10g)聚(δ-己内酯)(实施例1中的样品)切成碎片和(100mg)辛酸亚锡加入到反应瓶中并搅拌混合物。使用减压蒸馏装置,将反应瓶加热到130℃并持续2h,接受瓶得到δ-己内酯(9.91g,产率:99%)。本体解聚回收得到的δ-己内酯与原始单体的1h nmr叠加谱图如图8所示。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1