一种Tn5转座酶及其制备方法与流程
一种tn5转座酶及其制备方法
技术领域
1.本发明涉及生物酶技术领域,具体为一种转座酶,尤其是一种tn5转座酶及其制备方法。
背景技术:
2.tn5转座子可在体外插入到含有靶dna的载体上,在没有镁离子孵育时,tn5转座子能够形成稳定的tn5转座体复合体,可以通过电击进入活细胞,转座体经细胞内镁离子活化后,就能随机插入到宿主的基因组dna中。
3.tn5转座酶识别tn5转座子序列的内端(inside end,ie)、外端(outside end,oe)和嵌合端(mosaic end,me)序列,含有me序列片段的体外转座效率最高。tn5转座子的插入位点具有很高的随机性,因此被广泛的用于体外转基因(外源基因整合到宿主细胞)和二代测序建库等领域。
4.但是,目前行业内的tn5转座酶普遍在打断dna序列时,有序列偏向性,且稳定性不高。所以如何构建和制备一种新型的tn5转座酶突变体,使tn5转座酶在插入dna序列时,无序列偏向性选择,而且稳定性更高,是本行业研究的热点和难点。
技术实现要素:
5.本发明的目的在于提供一种tn5转座酶及其制备方法,以解决上述背景技术中提出的问题。
6.tn5转座酶的酶活单位定义:1单位tn5转座酶指37℃条件下反应1小时,完全切割1ug含有识别序列的dna片段所需要的酶量。
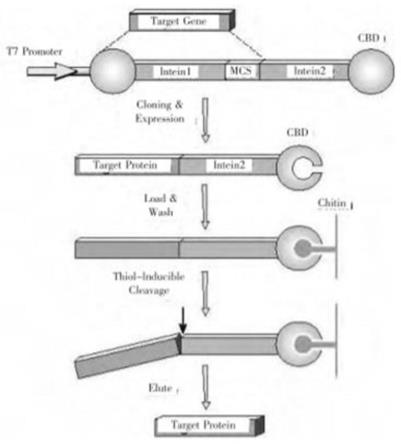
7.一种tn5转座酶,表达基因包括tn5转座酶基因、内含肽基因和几丁质基因。
8.优选的,所述表达基因序列如seq id no.1所示。
9.优选的,所述表达基因表达的氨基酸序列如seq id no.2所示。
10.本发明通过构建包含tn5转座酶基因、内含肽基因、几丁质基因的全长目的基因序列,将其连接到载体后导入大肠杆菌表达,采用一种新型的蛋白融合表达及纯化系统,得到的纯化tn5转座酶,可以高效的将tn5转座子插入到目标序列。
11.新型的蛋白融合表达及纯化系统(impact),impact表达的目的蛋白与内含肽(intein)及几丁质结合蛋白(cbd)形成融合蛋白,通过几丁质柱亲和纯化融合蛋白,dtt诱导内含肽的肽键裂解活性,在几丁质介质上将目的蛋白释放出来,而内含肽与几丁质结合蛋白仍结合在几丁质介质上,达到单柱分离纯化蛋白的目的,该方法具备比以往的亲和层析纯化方法更加经济的特点,方法更加新颖,可提高目的蛋白表达的成功率。
12.具体制备过程如下:
13.1、构建载体,种子培养,得到含目的基因的质粒;2、诱导表达:将目的基因的质粒转入大肠杆菌中表达;3、菌体富集处理,破碎大肠杆菌,得到粗酶裂解液;4、疏水层析,去除疏水性差异大的杂质;5、几丁质(chitin)亲和层析,捕获tn5、几丁质、内含肽的融合蛋白;
6、采用dtt切断内含肽,剔除几丁质标签,得到粗的tn5转座酶;7、采用离子交换层析对tn5进行精纯;8、采用凝胶过滤层析将tn5转座酶置换到保存液中;9、通过超滤浓缩得到高纯度高浓度的tn5转座酶。
14.优选的,所述质粒载体为pet-28a,所述rbs如seq id no.3所示。
15.本发明的制备原理如下:
16.1)按照seq id no.1合成全长基因序列
17.2)将拼接的全长基因序列xho i和xba i双酶切,通过t4连接酶克隆在pet-28a载体上。
18.3)连接产物转化dh5α感受态细胞中通过涂布硫酸卡那霉素抗性的平板培养。
19.4)通过pcr筛选阳性克隆。并通过测序验证突变体。
20.5)将筛选好的阳性克隆转化rosetta感受态细胞,诱导表达。
21.6)大肠杆菌湿菌体破碎诱导后的细胞,通过亲和层析色谱、几丁质树脂和离子交换色谱等纯化目的蛋白。
22.7)对突变体活性进行检测。
23.本发明通过impact系统表达的tn5转座酶,增加了tn5转座酶的无序列偏向性和热稳定性。
24.与现有技术相比,本发明所达到的有益效果是:
25.1、相较于其他的tn5转座酶及其制备方法,通过本发明的方法获得的tn5转座酶的纯度和浓度更高。
26.2、采用本发明提供的新型的蛋白融合表达及纯化系统(impact),简化了常规的繁琐提取过程,获得的tn5转座酶可以高效的将tn5转座子插入到目标序列、具有无序列偏向性,表现对目的片段(300-600bp)分选更集中、tn5转座子插入到目标序列的稳定性更高。
附图说明
27.附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
28.图1是impact的工作原理示意图;
29.图2是本发明的tn5转座酶的制备过程流程示意图;
30.图3是目的基因的质粒图谱tn5转座酶序列信息;
31.图4是实施例4中制备的tn5转座酶疏水层析色谱图一;
32.图5是实施例4中制备的tn5转座酶疏水层析色谱图二;
33.图6是实施例4中制备的tn5转座酶疏水层析电泳图一;
34.图7是实施例4中制备的tn5转座酶疏水层析电泳图二;
35.图8是实施例5中制备的tn5转座酶ni柱层析色谱图一;
36.图9是实施例5中制备的tn5转座酶ni柱层析色谱图二;
37.图10是实施例5中制备的tn5转座酶ni柱层析电泳图一;
38.图11是实施例5中制备的tn5转座酶ni柱层析电泳图二;
39.图12是实施例6的chitin亲和的蛋白电泳图一;
40.图13是实施例6的chitin亲和的蛋白电泳图二;
41.图14是实施例7的获得的tn5转座酶离子交换层析图;
42.图15是实施例7的获得的tn5转座酶离子交换电泳图一;
43.图16是实施例7的获得的tn5转座酶离子交换电泳图二;
44.图17是实施例7的获得的tn5转座酶离子交换电泳图三;
45.图18是实施例7的获得的tn5转座酶离子交换电泳图四;
46.图19是实施例8的获得的tn5转座酶凝胶过滤层析图一;
47.图20是实施例8的获得的tn5转座酶凝胶过滤层析图二;
48.图21是实施例8的获得的tn5转座酶离子交换电泳图一;
49.图22是实施例8的获得的tn5转座酶离子交换电泳图二;
50.图23是实施例8的获得的tn5转座酶离子交换电泳图三;
51.图24是实施例8的获得的tn5转座酶离子交换电泳图四;
52.图25是实施例9的获得的tn5转座酶离子交换电泳图一;
53.图26是实施例9的获得的tn5转座酶离子交换电泳图二;
54.图27是实施例9的获得的tn5转座酶离子交换电泳图三;
55.图28是实施例10的获得的tn5转座酶离子交换电泳图一;
56.图29是实施例12的50ul菌液涂布平板图;
57.图30是实施例12的100ul菌液涂布平板图;
58.图31是实施例12的获得的tn5转座酶离子交换电泳图一;
59.图32是实施例13的获得的tn5转座酶离子交换电泳图二;
60.图33是实施例13的获得的tn5转座酶离子交换电泳图三;
61.图34是实施例13的获得的tn5转座酶离子交换电泳图四;
62.图35是实施例13的获得的tn5转座酶离子交换电泳图五;
63.图36是实施例14的获得的tn5转座酶片段产物示意图一;
64.图37是实施例14的获得的tn5转座酶片段产物示意图二;
65.图38是实施例14的获得的tn5转座酶片段产物示意图三;
66.图39是实施例14的获得的tn5转座酶片段产物示意图四;
67.图40是实施例14的获得的tn5转座酶片段产物示意图五;
68.图41是实施例14的获得的tn5转座酶片段产物示意图六。
具体实施方式
69.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
70.实施例中的译酶为苏州译酶生物科技有限公司的简称。
71.实施例1:
72.构建载体
73.根据seq id no:1合成基因序列并将全基因片段插入载体pet-28a(xho i/xba i)载体中。
74.本实施例中使用的是pet-28a表达载体,但不限于此载体。
75.目的基因的质粒图谱tn5转座酶序列信息:
76.表达载体:pet-28a
77.酶切位点:xho i/xba i
78.启动子:t7
79.rbs序列:
80.tctagaaataattttgtttaactttaagaaggagatttaaat
81.(非pet-28a原有的rbs序列)
82.纯化标签:c端cbd标签
83.c端10个his标签。
84.rbs序列对原核基因的表达至关重要,如果mrna序列中的cds序列与rbs序列之间形成了强烈的二级结构,将会非常严重的影响翻译过程,从而造成基因表达量与预期差异巨大。因此,在标准化的原核转录单元序列设计中,应该考虑到rbs的因素,推荐大家使用rbs calculator计算rbs强度,从而来合理控制所需基因的表达水平。
85.本实施例1中的rbs序列与seq id no:1合成基因序列相匹配,提高了表达融合蛋白的稳定性,保证了本发明中所需基因的表达水平。
86.实施例2:
87.诱导表达
88.tn5转座酶突变体转化实验
89.取1支rosetta感受态(货号:cw0811s,批号:40426)细胞冰上放置20min,待感受态细胞解冻为液体状态;
90.取1μl 10ng/μl的tn5-pet 28a质粒加入到解冻的感受态细胞中(50-100μl),轻轻弹两下混匀,冰上静止25min;
91.42℃水浴热激90s后迅速冰浴5min;
92.加入1000μl新鲜无抗lb液体培养基(批号20200304#1),置于摇床200rpm 37℃培养1h;
93.kan+抗性(批号20200304#1)(浓度为0.5%):取200μl均匀涂布于kan+的lb平板中(0.5%),37℃恒温培养箱培养16h。
94.诱导表达
95.挑取上述平板中抗性高的长势正常的单克隆菌落到150ml kan+的lb液体培养基中(100ug/ml kan+,原液50mg/ml),置于摇床200rpm 37℃培养16h。
96.取10ml步骤培养好的菌种接种于1l的kan+的tb液体培养基中(100ug/ml kan+),200rpm 37℃培养摇床培养4h左右。
97.待培养的菌液浓度od600在0.8-1.0左右时,温度降到25℃,加入iptg终浓度为0.5、1.0mm,等加完iptg后,开始计时诱导表达16h。
98.使用落地离心机收菌,设置6500rpm,4℃,15min,去尽残留培养基收集沉淀,称量所有菌体湿重并记录,所有菌体置于零下20摄氏度保存。
99.实施例3:
100.菌体处理
101.高压破碎菌体,使用电子天平称量湿菌体40g,加入50ml tn5 ni buffer a(20mm hepes,800mm nacl,10%glycerol,0.2%tritonx-100,ph=7.2,25℃),使用涡旋混匀仪快速涡旋均匀直至肉眼观察无明显菌块,50ml离心管定容至400ml,置于冰浴中备用。
102.高压细胞破碎仪冷阱提前循环预冷至4℃,排尽管路中75%乙醇,加入500ml共艺用水清洗管路,然后加入200ml tn5 ni buffer a润洗管路后排空,倒入重悬菌液,1200bar,流速调至50,破碎6次,细胞破碎液置于冰浴暂存。泄压后加入500ml纯水冲洗管路,加入200ml75%乙醇冲洗管路,最后保证整个管路浸泡于75%乙醇。
103.破碎液处理
104.离心:将破碎液转移至离心管中,两两配平,15000rpm、4℃离心30min。分离沉淀和上清。
105.过滤:将上清转移至干净容器中,分别依次用0.45um、0.22um滤膜过滤,4℃待用。
106.pei沉淀
107.向离心后得到的补足至400ml上清中,边磁力搅拌边加入10%pei母液(批号2021009#1),直至加入的pei终浓度为0.4%为止,共计加入16ml,注意缓慢加入,防止目的蛋白沉淀;
108.室温磁力搅拌平衡10min,然后置于4℃放置20min;
109.15000rpm,4℃,离心30min,取上清,补体积至400ml;
110.加入3m(nh4)2so4补盐;
111.使用电导率仪测试上清,记录电导率数值。
112.加入3m(nh4)2so4母液,边磁力搅拌边加入硫(少多次加入上清中),直至电导显示为100
±
5ms/cm左右。记录加入3m(nh4)2so4的体积,待完全加入后室温搅拌平衡10min。
113.0.22μm水相微孔滤膜过滤,量取上清液体积,准备上样。
114.实施例4:
115.疏水色谱层析
116.疏水柱纯化(capto butyl,xk26/200mm,cv=50ml,流速8ml/min)
117.tn5 capto butyl buffer a 20mm hepes,1.2m(nh4)2so4,ph7.2,25℃
118.tn5 capto butyl buffer b 20mm hepes,ph7.2,25℃
119.管道清洗
120.对工艺用水、tn5 capto butyl buffer a、tn5 capto butyl buffer b分别超声脱气5分钟处理。
121.将akta的a1、a2、b1、b2、s1和buffer泵头放入工艺用水中,设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
122.系统泵及buffer放置。
123.将akta的a1、a2、b1、b2、s1和buffer泵放入下表buffer中
124.pumpmoving phasesystem pump a1tn5 capto butyl buffer asystem pump a2工艺用水system pump b1tn5 capto butyl buffer bsystem pump b20.2m naoh
sample pump buffertn5 capto butyl buffer asample pump s1tn5 capto butyl buffer a
125.设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
126.设置a2泵流速0.5ml/min,连接层析柱。
127.上样纯化
128.样品:补盐过滤后上清液,记录体积。
129.方法设置:
[0130][0131][0132]
管道和纯化柱清洗:待实验结束后,将层析柱依次使用20%乙醇清洗,将层析柱及系统管路保存在20%乙醇中,拆下层析柱,连接akta管路,将层析柱4℃保存。
[0133]
实施例5:
[0134]
亲和色谱层析:ni-hp柱纯化(ni-hp,流速5ml/min)
[0135]
填料:histrap ff 5ml
[0136]
柱体积:10ml
[0137]
上样体积:160ml
[0138]
系统流速:5ml/min
[0139]
缓冲液:
[0140]
tn5 ni buffer a:20mm hepes,800mm kcl,10%glycerol,0.2%tritonx-100,ph=7.2,25℃
[0141]
tn5 ni wash buffer:20mm hepes,800mm kcl,10%glycerol,2%tritonx-100,ph=7.2,25℃
[0142]
tn5 ni buffer b:20mm hepes,800mm kcl,10%glycerol,0.2%tritonx-100,250mm imidazole),ph=7.2,25℃
[0143]
管道清洗
[0144]
对工艺用水、tn5 ni buffer a、tn5 ni wash buffer、tn5 ni buffer b分别超声5分钟脱气处理。
[0145]
将akta的a1、a2、b1、b2、s1和buffer泵头放入工艺用水中,设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
[0146]
系统泵及buffer放置
[0147]
将akta的a1、a2、b1、b2、s1和buffer泵头放入下表buffer中
[0148]
pumpmoving phasesystem pump a1tn5 ni buffer asystem pump a2tn5 ni wash buffersystem pump b1tn5 ni buffer bsystem pump b2工艺用水sample pump buffertn5 ni buffer asample pump s1tn5 ni buffer a/sample
[0149]
设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
[0150]
设置b2泵流速0.5ml/min,连接层析柱。
[0151]
上样纯化
[0152]
样品:疏水层析后的目的蛋白洗脱液,记录上样体积。
[0153]
方法设置:
[0154][0155]
备注:第一个ni目的蛋白洗脱峰不收集,只收集第二个目的蛋白洗脱峰。
[0156]
管道和纯化柱清洗:待实验结束后,将层析柱依次使用500mm imidazole、工艺用
水、20%乙醇清洗,将层析柱及系统管路保存在20%乙醇中,拆下层析柱,连接akta管路,将层析柱4℃保存。
[0157]
实施例6:
[0158]
亲和色谱层析:chitin亲和纯化(chitin resin x 2,cv=20ml*2,填料载量:2mg/ml)
[0159]
1.层析柱:chromatography columns
[0160]
(bio-rad#7372507)1cv=10ml(填料)
[0161]
2.层析填料:chitin resin(neb#s6651l)
[0162]
3.buffer
[0163]
tn5 chitin ac buffer a:20mm hepes,800mm kcl,1mm edta,10%glycerol,0.2%tritonx-100ph=7.2,25℃
[0164]
tn5 chitin ac buffer b:20mm hepes,800mm kcl,1mm edta,10%glycerol,0.2%tritonx-100
[0165]
100mm dtt,ph=7.2,25℃
[0166]
上样量:20ml纯化柱分别上样20g湿菌获得样品,100ml,chitin resin x 2,总上样量100ml*2
[0167]
dtt切断内含肽处理时间:64h
[0168]
方法设置:(目的蛋白在elution 3里面)
[0169][0170][0171]
chitin亲和的蛋白电泳图如图12和图13所示:
[0172]
tn5转座酶大小约为60kda,tn5-intein-his融合蛋白大小约为80kda。
[0173]
实施例7:
[0174]
离子交换色谱层析:cm-ff柱纯化(cm-ff)
[0175]
1.层析柱:hitrap cm ff,5ml(ge,#17515501)
[0176]
2.buffer:
[0177]
tn5 iex buffer a:20mm hepes,20mm nacl,1mm edta,5%glycerol,0.05%tween-20,1mm dtt,ph=7.2,25℃
[0178]
tn5 iex wash buffer:20mm hepes,20mm nacl,1mm edta,5%glycerol,1%tritonx-100,1mm dtt,ph=7.2,25℃
[0179]
tn5 iex buffer b:20mm hepes,500mm nacl,1mm edta,5%glycerol,0.05%tween-20,1mm dtt,ph=7.2,25℃
[0180]
管道清洗
[0181]
对工艺用水、tn5 iex buffer a、tn5 wash buffer、tn5 iex buffer b分别超声5分钟脱气处理。
[0182]
将akta的a1、a2、b1、b2、s1和buffer泵头放入共艺用水中,设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
[0183]
系统泵及buffer放置
[0184]
将akta的a1、a2、b1、b2、s1和buffer泵头放入下表buffer中
[0185]
pumpmoving phasesystem pump a1tn5 iex buffer asystem pump a2tn5 wash buffersystem pump b1tn5 iex buffer bsystem pump b2工艺用水sample pump buffertn5 iex buffer asample pump s1tn5 iex buffer a/sample
[0186]
设置pump b2、a2、b1、s1、buffer和a1 pump wash,并执行。
[0187]
设置b2泵流速0.5ml/min,连接层析柱,使用水将乙醇冲出。
[0188]
上样纯化
[0189]
样品:chitin柱后洗脱液,使用tn5 iex dilute buffer(5mm hepes,2.5%glycerol,ph=7.2,25℃)稀释至电导值3ms/cm以下,进行离子交换层析。
[0190][0191]
方法设置:
[0192]
管道和纯化柱清洗:待实验结束后,将层析柱依次使用3m kcl,工艺用水,0.2mnaoh,工艺用水,20%乙醇清洗,然后将层析柱及系统管路保存在20%乙醇中,拆下层析柱,连接akta管路,将层析柱4℃保存。
[0193]
实施例8:
[0194]
凝胶过滤色谱层析:(hiload 16/600superdex 200pg,16mm x 700mm,cv=120ml)
[0195]
1.填料:hiload 16/600superdex 200pg
[0196]
柱体积:120ml
[0197]
流速:1ml/min
[0198]
上样量:1ml
[0199]
缓冲液:
[0200]
tn5 gel buffer:100mm hepes,200mm nacl,0.2mm edta,5%glycerol,2mm dtt,ph=7.4,25℃
[0201]
管道清洗
[0202]
对h2o、tn5 gel buffer分别超声5分钟脱气处理。
[0203]
将akta的a1、a2、b1、b2、s1和buffer泵头放入depc h2o中,设置pump b2、b1、a2、s1、buffer和a1 pump wash,并执行。
[0204]
系统泵及buffer放置
[0205]
将akta的a1、b1、a2、b2、s1和buffer泵头放入下表buffer中
[0206]
pumpmoving phasesystem pump a1tn5 gel buffersample pump buffertn5 gel buffersample pump s1tn5 gel buffer/sample
system pump b1h2osystem pump a20.2m naohsystem pump b220%乙醇
[0207]
设置pump b1、s1、buffer和a1 pump wash,并执行。
[0208]
设置b1泵流速0.5ml/min,连接层析柱。
[0209]
上样纯化(上样体积:2%-3%of the cv,0.22μm滤膜过滤或10000g离心10min,conc.《10mg/ml)。
[0210]
样品:cm柱后目的洗脱液,使用uv280测试浓度,消光系数为k=1.475,记录浓度和体积。使用30kd的超滤管对蛋白洗脱液进行离心(3500rpm,4℃),使浓度小于10mg/ml,记录浓缩后的总体积,可使用定量环分批上样,每批上样量为1-2ml。
[0211]
方法设置:
[0212][0213][0214]
管道和纯化柱清洗:待实验结束后,将层析柱及系统管路保存在20%乙醇中,拆下层析柱,连接akta管路,将层析柱4℃保存。
[0215]
备注:如果柱前压力≤0.8mpa,可以将流速调整为1.5ml/min。
[0216]
实施例9:
[0217]
膜包浓缩
[0218]
使用uv280测试蛋白浓度,消光系数k=1.475定量上述步骤收集的蛋白浓度mg/ml,分别记录体积和目的蛋白的量;
[0219]
清洗膜包:将膜包连接至蠕动泵上,设置蠕动泵转速300rpm,将进液管放入超纯水中,出液管放入废液桶中,开始清洗膜包,清洗体积约200ml后清空膜包中的液体;
[0220]
蛋白浓缩:将进液管、出液管同时放入待浓缩中,启动蠕动泵开始浓缩,浓缩至“tn5-gel”体积为膜包最小体积,然后清空膜包及管道内酶液;
[0221]
膜包浓缩后,经0.22um滤膜过滤后,使用uv280测试蛋白浓度,消光系数k=1.475定量过滤的蛋白浓度=1.0mg/ml;
[0222]
加入等体积灭菌甘油,记录批号和加入体积(ml),浓度为0.50mg/ml;
[0223]
补加添加剂:
[0224]
终浓度为0.1%triton-100,记录100%triton-100母液批号(批号:slbw7103)和加入体积(μl);
[0225]
终浓度为0.1%tween-20,记录100%tween-20母液批号(批号:slbz8563)和加入体积(μl);
[0226]
将高浓度成品tn5,记录批号,浓度为0.50mg/ml),母液分成500μl/tube,-20℃保存。
[0227]
实施例10:
[0228]
tn5转座酶定量和纯度检测
[0229]
检测过程:使用uv280测试蛋白浓度,消光系数k=1.475定量过滤的蛋白浓度=1.0mg/ml;
[0230]
加入等体积灭菌甘油,记录批号(批号:20181212)和加入体积(ml),浓度为0.50mg/ml;
[0231]
20200304#1p1经过浓缩后sds-page分析纯度为低于80%左右,纯度较低。
[0232]
电泳图见图25。
[0233] 123来源译酶tn5mark标志物强微特上样体积5ul3ul2ul泳道酶量3.7ug-预估为1.23ug
[0234]
12g菌体(1l菌液)可纯化出10mg蛋白
[0235]
pierce
tm bca protein assay kit
[0236]
(thermo scientific,lot:23227)
[0237]
分离胶浓度:10%
[0238]
单孔上样体积:5ul
[0239][0240]
电泳图见图26、图27,
[0241]
20200313#1p1经过浓缩后sds-page分析纯度为89.3%,
[0242]
20200313#1p2经过浓缩后sds-page分析纯度为92.8%。
[0243]
实施例11:
[0244]
tn5转座酶内切酶残留检测
[0245]
依据国标gb/t 35542-2017进行检测。
[0246]
puc19质粒反应
[0247]
在测试管里先后加入4ul puc19质粒dna(1ug/ul)、14ul纯化水、2ul酶,振荡器混匀,水浴1h;
[0248]
在对照管里先后加入4ul puc19质粒dna(1ug/ul)、16ul纯化水,振荡器混匀,水浴1h;
[0249]
电泳1%—1.5%琼脂糖凝胶,电泳电压5v/cm—10v/cm,当溴酚蓝线到达凝胶板2/3时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照;
[0250][0251]
37℃反应1h后,去20ul样品-80℃冷存,剩余样品(约30ul)37℃反应14h,电泳检测,详见图28。
[0252][0253][0254]
实施例12:
[0255]
tn5转座酶外切酶残留检测
[0256]
检测方法依据国标gb/t 35542-2017进行检测
[0257]
长时间酶切:37℃反应10h后,
[0258][0259]
37℃反应10h后,取样进行琼脂糖凝胶电泳检测
[0260]
dna回收
[0261]
取长时间酶切产物20ul分别加入20ul氯仿-异戊醇溶液(氯仿:异戊醇(v/v)=24:1)洗脱杂白质,沉淀dna,dna进行真空冷冻干燥。
[0262]
连接反应
[0263]
项目添加量真空干燥样品0ult4 dna ligase1ul10*t4 buffer2ulddh2o17ul总体积20ul
[0264]
15℃反应8h后,取2ul连接产物转化到dh5α细胞中,按不同梯度涂布平板,37℃培养过夜。
[0265][0266]
实验组(2,3,4)的白斑总数分别比上个对照组(1)的蓝斑总数,比值均小于1/9,详见于图29、图30。
[0267][0268][0269]
电泳图于图31。
[0270]
实施例13:
[0271]
该突变体相比于野生型,表现对目的片段(300-600bp)分选更集中。
[0272]
本实施例13中的tn5转座酶突变体是指经过实施例1-12过程制备的tn5转座酶,提到的商品酶或者tn5转座酶野生型均为强微特的tn5转座酶(型号m0221)
[0273]
tn5meds制备
[0274]
将tn5me-a、tn5me-b和tn5merev 12000rpm离心1min,加入1 x te buffer将其稀释至100um。
[0275]
按照下表配制体系,运行热循环程序:
[0276]
componentsconcentrationtn5me-a(100μm)50μltn5merev(100μm)50μl
[0277]
componentsconcentretiontn5me-b(100μm)50μltn5merev(100μm)50μl
[0278][0279]
transposome合成
[0280][0281]
pcr仪热盖60℃,25℃,1h,-20℃分保存。
[0282]
gdna片段化和纯化(50ng)
[0283]
componentsconcentrationk562gdna(qubit 10ng/μl)5μl5x lm buffer/5xtamd buffer6μltransposome1ul/2μldepc waterup to 30μl
[0284]
pcr仪预热55℃,热盖105℃。
[0285]
55℃片段化10min,立即取出冰浴降温3min,加入1ul 5%sds混合均匀。
[0286]
55℃,5min终止反应,立即取出冰浴降温3min。
[0287]
使用1 x kapapure beads纯化回收片段,23ul depc water洗脱。
[0288]
cdna片段化和纯化(5ng)
[0289]
componentsconcentrationk562cdna(qubit 4ng/μl)125μl5x lm buffer/5xtamd buffer6ultransposome1ul/2μldepc waterup to 30μl
[0290]
pcr仪预热55℃,热盖105℃。
[0291]
55℃片段化10min,立即取出冰浴降温3min,加入1ul 5%sds混合均匀。
[0292]
55℃,5min终止反应,立即取出冰浴降温3min。
[0293]
使用1 x kapapure beads纯化回收片段,23ul depc water洗脱。
[0294]
pcr富集与片段分选。
[0295]
componentsconcentrationn5xx(10μm)2uln7xx(10μm)2μl2
×
kapa hifi hotstar ready mix25μldna21μl
[0296][0297]
pcr富集结束,使用0.6x/0.15x kapapure beads分选300-600bd片段,26ul depc water洗脱。
[0298]
gdna人类基因组dna片段化、pcr富集和300bp-600bp分选:
[0299]
实验结论:55℃,10min片段化:20200313#1p1 tn5转座酶突变体片段化效果接近商品酶,且同样transposome转座子添加量下自产tn5转座酶脱落adapter连接体均少于商品酶,但20200313#1p1和20200313#1p2两组副带差异tn5转座酶,片段化效果差异不明显,详见于图32。
[0300]
pcr富集和300bp-600bp分选:经过pcr富集译酶tn5片段文库大小与强微特组差异不明显,经过300-600bp片段分选后,所有实验组均无300bp以下小片段。20200313#1p1tn5转座酶突变体转座酶性能优于20200304#1p1野生型tn5,详见于图33。
[0301]
30ul片段化体系添加1ul transposome组中强微特组和20200313#1p1 tn5转座酶突变体组片段化完全,20200304#1p1 tn5野生型组明显未片段化完全。
[0302]
30ul片段化体系添加2ul transposome组中强微特组和20200313#1p1 tn5转座酶突变体组片段化完全,20200304#1p1 tn5野生型组明显未片段化完全。
[0303]
总体分析,20200313#1p1 tn5转座酶突变体片段化效果接近商品酶,且同样transposome添加量下自产tn5转座酶脱落adapter均少于商品酶,但20200313#1p1和20200313#1p2两组副带差异tn5转座酶,片段化效果差异不明显。
[0304]
pcr富集和300-600bp片段分选:
[0305]
经过pcr富集译酶tn5片段文库大小与强微特组差异不明显,经过300-600bp片段分选后,所有实验组均无300bp以下小片段。
[0306]
30ul片段化体系添加1ul transposome组中均存在1000bp以上大片段,且3组自产tn5转座酶大片段与商品酶差异不明显。
[0307]
本组20200313#1p1和20200313#1p2两组副带差异tn5转座酶,pcr富集和片段化后分选差异不明显。
[0308]
30ul片段化体系添加2ul transposome组20200313#1p1组片段效果和大片段残留量最接近商品酶。本组20200313#1p1和20200313#1p2两组副带差异tn5转座酶,20200313#1p1tn5转座酶(副带少)效果最接近商品酶,大片段残留量少于20200313#1p2(副带多)。
[0309]
总体可看出20200313#1tn5转座酶突变体性能优于20200304#1p1 tn5转座酶野生型。
[0310]
cdna片段化、pcr富集和300bp-600bp分选:
[0311]
实验结论:55℃,10min片段化:在同样transposome转座子添加量下自产tn5转座
酶脱落adapter均少于商品酶。详见于图34。
[0312]
pcr富集和300bp-600bp分选:20200313#1p1 tn5转座酶组和商品酶组富集后文库大小等总体差异不明显。详见于图35。
[0313]
明显可见在同样transposome添加量下自产tn5转座酶脱落adapter均少于商品酶。
[0314]
pcr富集和300-600bp片段分选:
[0315]
20200304#1p1 tn5转座酶野生组残留的dimer也更重,20200313#1tn5转座酶突变体组和商品酶组富集后文库大小等总体差异不明显。
[0316]
经过pcr富集译酶tn5片段文库大小与强微特组差异不明显,经过300-600bp片段分选后,所有实验组均无300bp以下小片段。
[0317]
30ul片段化体系添加1ul transposome组文库量和纯净度优于30ul片段化体系添加2ul transposome组,推断为起始模板量少加入transposome过多导致片段化过碎产生小片段,导致所需目的片段(300bp-600bp)降低。
[0318]
同样transposome添加量下,20200313#1tn5转座酶突变体组和商品酶组差异不明显。
[0319]
总体可看出20200313#1tn5转座酶突变体性能优于20200304#1p1 tn5转座酶野生组。
[0320]
实施例14:
[0321]
tn5转座酶该突变体具有无序列偏向性
[0322]
使用kapa酶切试剂盒(kapa货号kk8600)进行片段化实验
[0323]
本实施例14中的tn5转座酶突变体是指经过实施例1-12过程制备的tn5转座酶,对照的野生型tn5转座酶均为强微特的tn5转座酶(型号m0221)
[0324]
实验结果如下:
[0325]
(1)根据图36可知,tn5转座酶突变体:批号:20200313#1p1,均一性比较好。
[0326]
(2)根据图37可知,tn5转座酶突变体:批号:20200313#1p2,均一性比较好。
[0327]
(3)根据图38、39可知,野生型tn5转座酶:批号:20200304#1p1,均一性较差。
[0328]
(4)根据图40、41可知,对照的试剂盒:切的片段大小不均或者片段没切开。
[0329]
综上可知:通过构建包含tn5转座酶基因、内含肽基因、几丁质基因的全长目的基因序列,将其连接到载体后导入大肠杆菌表达,采用一种新型的蛋白融合表达及纯化系统,得到的纯化tn5转座酶,可以高效的将tn5转座子插入到目标序列,而且无序列偏向性选择。
[0330]
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
[0331]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。
凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
[0332]
[0333]
[0334]
[0335]
[0336]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1