一种用于同时检测金刚烷胺、喹乙醇、氯霉素的半抗原、人工抗原及其制备方法和应用
1.本发明涉及抗原抗体检测技术领域,更具体地,涉及一种同时含金刚烷胺 (amd)、3-甲基-喹恶啉-2-羧酸(mqca)及氯霉素胺(capa)骨架结构的 amc型多簇串联线性半抗原、人工抗原及其制备方法和应用。
背景技术:
2.我国是世界上最大的畜产品生产和消费大国,据统计,2010年我国肉类总 产量己达7925.8万吨,居世界首位。目前已报道的畜产品兽药残留分析检测技 术主要集中大型精密仪器分析技术和免疫分析筛查技术,其中仪器确证法以液相 色谱-串联质谱法(hplc-ms/ms)为主。例如中国专利公开了一种用于测定中 兽药散剂中11种非法添加药物含量的方法,可同时检测包含金刚烷胺、喹乙醇、 氯霉素等在内的11种非法添加药物进行检测,然而仪器法通常具有样品预处理 操作复杂、费时、成本高,不适合现场检测等缺点。免疫分析法具有灵敏度与常 规的仪器分析一致,适合现场筛选,简单、快速、成本低、样品所需量少、前处 理简单方便等优点。目前,畜产品安全检测中以单残留免疫检测方法为主,例如 中国专利cn108152499a、cn111077319a、cn108519480a等分别公开了针对 金刚烷胺、喹乙醇、氯霉素的免疫检测方法。然而,由于兽药种类的多样性及其 复配使用的广泛性和复杂性,仅发展单残留免疫检测技术已不能满足实际检测需 求,但是目前缺少用于同时检测非共有结构小分子药物金刚烷胺、喹乙醇、氯霉 素的相关半抗原、抗原和抗体。
3.
技术实现要素:
4.本发明要解决的技术问题是克服现有技术中存在的上述缺陷和不足,提供一 种可以实现非共有结构小分子药物金刚烷胺、喹乙醇、氯霉素的多残留免疫检测 的半抗原。通过将金刚烷胺、喹乙醇、氯霉素这三个不同结构小分子药物视为不 同表位,将其通过适当方式串联成线性多簇半抗原,以此来提高多表位中和识别 能力。
5.本发明的第二个目的是提供所述半抗原的制备方法。
6.本发明的第三个目的是提供所述半抗原的应用。
7.本发明的第四个目的是提供一种人工抗原。
8.本发明的第五个目的在于提供所述人工抗原的制备方法。
9.本发明的第六个目的在于提供所述人工抗原的应用。
10.本发明的第七个目的在于提供一种抗原特异性抗体。
11.本发明的上述目的通过以下技术方案予以实现:
12.一种用于同时检测金刚烷胺、喹乙醇、氯霉素的半抗原,同时含金刚烷胺 (amd)、3-甲基-喹恶啉-2-羧酸(mqca)及氯霉素胺(capa)骨架结构,其 结构通式如式ⅰ所示:
[0013][0014]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即r1和r2分别选自乙 基至戊基中的任意一种。
[0015]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0016]
进一步优选地,所述半抗原结构式如下amc-4c或amc-6c所示:
[0017][0018][0019]
所述性半抗原的制备方法,包括如下步骤:
[0020]
s1.分别制备喹乙醇代谢物、金刚烷胺、氯霉素中间体:以乙酰乙酸甲酯为 原料经过nbs溴代、4-硝基邻苯二胺环化、还原硝基、上氨基保护基、酯键水 解,得到式ⅱ所示喹乙醇代谢物中间体;以金刚烷为原料经过硝化、硝基还原、 氨基酰化、水解后在其中1个氨基上连接叔丁氧羰基保护的氨基酸手臂,然后, 在另1个氨基上连接芴甲氧羰酰基,最后脱除boc保护基,得到式ⅲ所示金刚烷 胺中间体;将氯霉素的二氯乙酰基水解除去,得到式ⅳ所示氯霉素中间体氯霉素 胺;
[0021][0022]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即r1和r2分别选自乙 基至戊基中的任意一种;
[0023]
s2.半抗原的合成:将式iii所示金刚烷胺中间体与式ii所示喹乙醇代谢物中 间
体,通过分子上的伯氨基与羧基缩合偶联,然后脱除喹乙醇代谢物中间体的芳 环氨基的dmb保护基后,通过酰化缩合反应偶联得到中间体
ⅴ
,然后将中间体
ꢀⅴ
通过其羧基与式ⅳ所示氯霉素中间体的氨基经酰化缩合得到中间体ⅵ,再进一 步脱去fmoc保护基后分别与4-氧丁酸甲基酯缩合还原胺化为中间体ⅶ,最后水 解酯基,得到式ⅰ所示的半抗原。
[0024]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0025]
本发明还提供所述半抗原在制备人工抗原中的应用。
[0026]
一种人工抗原,由上述式ⅰ所示半抗原与载体蛋白偶联得到,其结构如式
ⅷꢀ
所示:
[0027][0028]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即所述r1和r2分别选 自乙基至戊基中的任意一种。
[0029]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0030]
进一步优选地,所述r1和r2分别选自丙基和乙基组合,戊基与丁基组合, 其取代后的结构式如下amc-4c-载体蛋白或amc-6c-载体蛋白所示:
[0031][0032]
优选地,所述载体蛋白为乳铁蛋白(lf)、钥孔血蓝蛋白(klh)或卵清蛋 白(ova)。lf、klh用于制备免疫原,ova用于制备包被原。
[0033]
所述人工抗原的制备方法,是在式ⅰ所示半抗原的羧基上偶联载体蛋白得到。
[0034]
优选地,所述偶联为通过活泼酯法。
[0035]
作为一种优选地可实施方式,所述人工抗原的制备方法,包括如下步骤:将 式ⅰ所示半抗原、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(edc)和n-羟基 丁二酰亚胺(nhs)溶于n,n-二甲基甲酰胺(dmf)中,4℃搅拌反应过夜,记 为a液。称取载体蛋白溶于磷酸缓冲溶液(pbs,0.01m,ph 7.4)中,搅拌溶 解制成b液。磁力搅拌下,吸取a液逐滴加入到b液中,4℃下磁力搅拌反应, 将反应液于4℃下用pbs透析3天,每天换2次透析液,即可得到人工抗原。
[0036]
本发明还提供所述人工抗原在制备单/多克隆抗体、单链抗体或纳米抗体中 的应用。
[0037]
一种多克隆抗体,是以式
ⅷ
所示人工抗原免疫小鼠制备得到。
[0038]
优选地,是以载体蛋白为乳铁蛋白(lf)或钥孔血蓝蛋白(klh)的人工 抗原为免疫原,选取适龄雌性balb/c小鼠进行免疫,第4次免疫和第5次免疫 后测定抗血清的效价和抑
制率,待性能稳定后收集小鼠血清,得到多克隆抗体。
[0039]
本发明还提供一种检测人工抗原的快速免疫分析方法,基于elisa方法进 行抗血清性能评价,以载体蛋白为卵清蛋白(ova)的人工抗原为包被原,用上 述抗原特异性抗体作为检测抗体进行检测。优选地,所述抗原特异性抗体为鼠多 克隆抗体。
[0040]
与现有技术相比,本发明具有以下有益效果:
[0041]
本发明首先提供了一种用于同时检测金刚烷胺、喹乙醇、氯霉素的半抗原, 其结构通式如式i所示,是将金刚烷胺、喹乙醇代谢物和氯霉素3种药物的中间 体与不同间隔臂的氨基羧酸分子连接,进行混合化学串联合成多簇串联线性半抗 原。基于该半抗原制备人工抗原,再以人工抗原免疫动物制备获得的鼠多抗血清, 效价在8~32k之间,对多簇串联线性半抗原的抑制率为63~90%。表明本发明制 备的人工抗原具有良好的免疫原性,可以用于后续制备多簇串联线性抗原或多表 位特异性单克隆抗体、单链抗体和纳米抗体并建立相对应免疫分析法。本发明为 小分子多特异性抗体制备及多残留分析方法奠定基础,具有良好的应用前景。
附图说明
[0042]
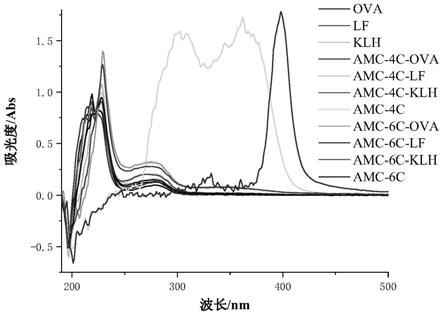
图1为多簇串联线性半抗原、人工抗原、载体蛋白紫外扫描图。
具体实施方式
[0043]
下面结合附图和具体实施例进一步详细说明本发明,除非特别说明,本发明 采用的试剂、方法和仪器为本技术领域常规的试剂、方法和仪器。
[0044]
实施例1amc型多簇串联线性半抗原的制备
[0045]
1、一种amc-4c半抗原的制备方法,包括如下步骤:
[0046]
s1.中间体3的制备
[0047][0048]
将乙酰乙酸甲酯即化合物1(50g,431mmol)于水(1l)中,在70℃下 加入n-溴代琥珀酰亚胺(即nbs)(88g,494mmol)并在室温下搅拌半小时至 体系透明。随后加入4-硝基邻苯二胺即化合物2(63g,412mmol)并在70℃下 继续搅拌8小时,经液-质联用质谱仪(即lc-ms)监测反应完全后,冷却至室 温,用pe:ea=1:1的有机溶剂萃取(500ml
×
6)。合并有机相,干燥,旋干。粗 产品经柱层析(pe:ea=10:1)分离纯化得中间体3(14g,收率:13.8%,57mmol) 的黄色固体,长时间旋干后变紫黑色固体。ms(esi)m/z(m+h)
+
248.1。
[0049]
s2.中间体4的制备
[0050][0051]
将中间体3(14g,57mmol)溶于四氢呋喃(即thf)(100ml)和甲醇(100 ml)中,加入zn(40g,634mmol)。在0℃下分批加入氯化铵(38g,710mmol) 并继续在0℃下搅拌反应半小
时,经液-质联用质谱仪(即lc-ms)监测反应完 全后,反应液通过硅藻土过滤,滤饼用二氯甲烷和甲醇的混合溶剂洗涤。滤液浓 缩。剩余物经柱层析(pe:ea=100:0-50:50)分离纯化得中间体4(7g,收率: 56.14%,32mmol)的黄色固体。ms(esi)m/z(m+h)
+
218.1。1h nmr(400mhz, dmso)δ7.72(d,j=9.0hz,1h),7.32(dd,j=9.0,2.5hz,1h),6.90(d,j=2.4hz, 1h),6.11(s,2h),3.93(s,3h),2.68(s,3h)。
[0052]
s3.中间体5的制备
[0053][0054]
在室温下,将中间体4(7g,32mmol)和2,4-二甲氧基苯甲醛(即dmb)(10.2 g,61.4mmol)溶于二氯甲烷(即dcm)(100ml)中,加入三乙酰氧基硼氢化钠 (即stab)(20.5g,96.7mmol)在室温下搅拌反应过夜。经液-质联用质谱仪(即 lc-ms)监测反应完全后,将水(100ml)加入反应液中,用ea(100ml
×
5) 萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫酸钠干燥过滤后浓缩。 剩余物经柱层析(pe:ea=100:0-65:35)分离纯化得中间体5(8.5g,收率:72.2%, 23.1mmol)的黄色固体。ms(esi)m/z(m+h)
+
368.0。1h nmr(400mhz,dmso) δ7.74(t,j=11.0hz,1h),7.43(dt,j=27.5,13.8hz,1h),7.17(d,j=8.3hz,1h), 7.05(t,j=5.6hz,1h),6.63(dd,j=16.4,2.4hz,2h),6.47(dd,j=8.3,2.4hz,1h), 4.28(d,j=5.6hz,2h),3.91(s,3h),3.85(s,3h),3.74(s,3h),2.69(s,3h)。
[0055]
s4.中间体6的制备
[0056][0057]
将中间体5(8.5g,23.1mmol)和naoh(1.85g,46.2mmol)溶于thf(20 ml)和水(10ml)中。反应液在室温下搅拌反应1小时。经液-质联用质谱仪 (即lc-ms)监测反应完全后,用浓盐酸调ph至弱酸性,随后用dcm(100ml
×
8) 萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫酸钠干燥。过滤后浓缩 得中间体6(7.4g,收率:90.5%,20.9mmol)的橙色固体,即喹乙醇代谢物(3
‑ꢀ
甲基-喹恶啉-2-羧酸)中间体。ms(esi)m/z(m+h)
+
354.1。1h nmr(400mhz, dmso)δ7.70(d,j=9.1hz,1h),7.40(dd,j=9.1,2.4hz,1h),7.17(d,j=8.3hz, 1h),7.00(s,1h),6.62(dd,j=16.2,2.4hz,2h),6.47(dd,j=8.4,2.3hz,1h),4.27 (d,j=5.6hz,2h),3.85(s,3h),3.73(s,4h),2.66(s,3h)。
[0058]
s5.中间体8的制备
[0059][0060]
将金刚烷胺,即化合物7(75g,551mmol)在0℃下缓慢加入搅拌的发烟 硝酸中(345g)中,并在0℃下继续搅拌反应1小时。随后在0℃下滴加乙腈(55.5 g,1.37mol)并继续
在0℃下搅拌1小时。最后在0℃下缓慢加入浓硫酸(705g), 随后在室温下搅拌过夜。经液-质联用质谱仪(即lc-ms)监测反应完全后,将 反应液倒入冰(大约1kg)中,用碳酸钠固体调节ph至碱性,有黄色粘稠状物 体析出。将水倾析出,加入ea(大约100ml)使黄色粘稠物质结块,过滤。黄 色沉淀用ea洗涤后用水洗涤得中间体8(80g)的白色固体。ms(esi)m/z(m+h) +
251.1。1h nmr(400mhz,dmso)δ7.36(s,2h),2.11(d,j=10.5hz,2h),2.10
–ꢀ
1.98(m,2h),1.91
–
1.74(m,8h),1.73(s,6h),1.49(s,2h)。
[0061]
s6.中间体9的制备
[0062][0063]
将中间体8(70g)于hcl(10%,500ml)中,在110℃下搅拌反应2天, 经液-质联用质谱仪(即lc-ms)监测反应完全后,将反应液减压浓缩得中间体 9(63g,264mmol)的白色固体。ms(esi)m/z(m+h)
+
167.1。1h nmr(400mhz, dmso)δ8.40(s,6h),2.33
–
2.20(m,2h),2.02(s,2h),1.84
–
1.66(m,8h),1.52(s, 2h).
[0064]
s7.中间体11的制备
[0065][0066]
将叔丁氧羰基(boc)保护的4-氨基丁酸,即化合物10(5.66g,27.8mmol)、 hatu(12.7g,33.4mmol)和diea(10.8g,83.6mmol)溶于dmso(50ml) 中并在室温下继续搅拌反应1小时。将上述液体缓慢滴加入正在搅拌的中间体9 (10g,41.8mmol)和diea(10.8g,83.6mmol)的dmso(100ml)溶液中, 并在室温下继续搅拌反应半小时,经液-质联用质谱仪(即lc-ms)监测反应完 全后,向反应液中直接加入fmoccl(21.63g,83.6mmol)和diea(10.8g,83.6 mmol),并继续搅拌反应1小时,经液-质联用质谱仪(即lc-ms)监测反应完 全后,加入水(300ml),用ea(100ml
×
5)萃取,有机层用饱和食盐水洗涤 (100ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经柱层析 (pe:ea=100:0-35:65)分离纯化得中间体11(6.9g,收率:43.2%,12mmol) 的黄色固体。ms(esi)m/z(m+h)
+
574.3。1h nmr(400mhz,dmso)δ7.88(d,j =7.5hz,1h),7.71(d,j=7.4hz,1h),7.48
–
7.36(m,1h),7.33(td,j=7.4,1.1hz, 1h),7.11(s,1h),6.76(s,1h),4.18(s,1h),2.94
–
2.79(m,1h),2.11(s,2h),2.03
–ꢀ
1.93(m,3h),1.92
–
1.61(m,4h),1.54(dt,j=14.3,7.2hz,2h),1.37(s,5h)。
[0067]
s8:中间体12的制备
[0068][0069]
将中间体11(6.9g,12mmol)溶于4m的盐酸二氧六环溶液中(50ml) 中并在室温下继续搅拌反应1小时。tlc显示反应完全。将反应液浓缩得中间 体12(7g)的淡黄色油状液体,即中间体12。ms(esi)m/z(m+h)
+
474.2。1h nmr(400mhz,dmso)δ7.89(d,j=7.5hz,6h),
ms)监测反应完全后,反应液过滤,用甲醇洗涤滤 饼后,滤液浓缩。剩余物经反相柱层析(fa,水:乙腈=100:0-40:60)分离纯化 得中间体16(1.4g,收率:50%,1.85mmol)的淡黄色固体。ms(esi)m/z(m+h) +
759.3。1h nmr(400mhz,dmso)δ10.50(s,1h),8.86(t,j=5.8hz,1h),8.57(d, j=2.2hz,1h),8.14(s,1h),7.98(d,j=9.1hz,1h),7.88(dd,j=9.1,2.3hz,3h), 7.71(d,j=7.4hz,2h),7.41(t,j=7.3hz,3h),7.33(td,j=7.4,1.1hz,2h),7.12(s, 1h),4.20(d,j=15.4hz,3h),2.79(s,3h),2.71
–
2.63(m,2h),2.58(t,j=6.8hz, 2h),2.20
–
2.05(m,6h),1.91
–
1.71(m,10h),1.57
–
1.41(m,3h)。
[0079]
s12.中间体17的制备
[0080][0081]
将氯霉素cap(25g,77.6mmol)溶于hcl(1m,500ml)中,在100℃ 下搅拌水解过夜,经液-质联用质谱仪(lc-ms)监测反应完全后,将反应液冷 却至室温,用乙酸乙酯ea(500ml
×
3)萃取除掉二氯乙酸。剩余水相旋干得白 色固体即中间体17(15g,收率:91.1%,70.7mmol)。
[0082]
s13.中间体18的制备
[0083][0084]
将中间体16(1.4g,1.85mmol)、中间体17(3.92g,18.5mmol)和diea(1.2 g,9.25mmol)溶于dmf(20ml)中,加入1-丙基磷酸三环酸酐即t3p(2.35g, 7.4mmol)并在室温下继续搅拌反应1小时,经液-质联用质谱仪(即lc-ms) 监测反应完全后,加入水(50ml),用ea(50ml
×
3)萃取,有机层用饱和食 盐水洗涤(50ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经反相柱(fa,水: 乙腈=100:0-50:50)分离纯化得中间体18(950mg,收率:54%,1.0mmol)的 淡黄色固体。ms(esi)m/z(m+h)
+
953.5。1h nmr(400mhz,dmso)δ10.38(s, 0h),8.84(t,j=5.8hz,0h),8.53(d,j=2.2hz,0h),8.12(d,j=8.8hz,1h),7.96 (d,j=9.1hz,0h),7.86(dd,j=13.6,4.6hz,1h),7.71(d,j=7.5hz,1h),7.60(s, 1h),7.59(d,j=8.7hz,1h),7.40(t,j=7.4hz,1h),7.35
–
7.27(m,1h),5.80(s, 0h),5.00(s,0h),4.18(s,1h),3.98(d,j=8.3hz,0h),3.61
–
3.53(m,1h),3.17(s, 2h),2.79(s,1h),2.44
–
2.35(m,1h),2.10(dd,j=13.6,6.2hz,2h),2.02
–
1.95(m, 1h),1.92
–
1.69(m,4h),1.49(s,1h)。
[0085]
s14.中间体19的制备
[0086][0087]
将中间体18(950mg,1.0mmol)溶于乙醇即etoh(20ml)中,加入二 乙胺(5ml)并在室温下继续搅拌反应6小时,经液-质联用质谱仪(即lc-ms) 监测反应完全后,将反应液浓缩,用etoh带干两次。剩余物用水(5ml)溶解, 用石油醚即pe洗去杂质后冻干得中间体19(720mg,收率:98.6%,0.98mmol) 的淡黄色固体。ms(esi)m/z(m+h)
+
731.1。1h nmr(400mhz,
dmso)δ10.43 (s,1h),8.83(t,j=5.8hz,1h),8.54(d,j=2.1hz,1h),8.12(d,j=8.7hz,2h), 7.96(d,j=9.0hz,1h),7.85(dd,j=9.1,2.2hz,1h),7.67(d,j=9.2hz,1h),7.59 (d,j=8.7hz,2h),7.37(s,1h),5.00(d,j=2.2hz,1h),3.98(d,j=8.4hz,1h), 3.58
–
3.48(m,6h),3.17(s,1h),2.80(s,4h),2.46
–
2.38(m,3h),2.15
–
1.92(m, 7h),1.79(t,j=21.7hz,11h),1.46(s,8h),1.21(d,j=19.9hz,6h)。
[0088]
s15.中间体21的制备
[0089][0090]
将中间体19(720mg,0.98mmol)、4-氧丁酸甲基酯即,化合物20(220mg, 1.88mmol)和stab(1.2g,5.64mmol)于dcm(20ml)和甲醇(20ml)中, 在室温下搅拌过夜,经液-质联用质谱仪(即lc-ms)监测反应完全后,反应液 旋干,剩余物经反相柱(氨水,水:乙腈=100:0-70:30)分离纯化得淡黄色固体即 中间体21(690mg,收率:84.7%,0.83mmol)。ms(esi)m/z(m+h)
+
831.2。 1
h nmr(400mhz,dmso)δ10.42(s,1h),8.83(d,j=5.5hz,1h),8.55(d,j=2.2 hz,1h),8.33(s,2h),8.13(d,j=8.8hz,2h),7.97(d,j=9.1hz,1h),7.86(dd,j= 9.1,2.3hz,1h),7.66(d,j=9.1hz,1h),7.59(d,j=8.7hz,2h),7.46(s,1h),5.33 (t,j=4.8hz,1h),5.01(s,1h),3.97(s,3h),3.57(d,j=2.2hz,4h),2.80(s,3h), 2.62(d,j=7.1hz,2h),2.38(t,j=7.4hz,4h),2.11(d,j=7.1hz,4h),2.00(dd,j =15.1,7.3hz,4h),1.91(s,2h),1.84(s,2h),1.80
–
1.72(m,5h),1.72
–
1.62(m, 3h),1.58(s,5h),1.49(d,j=10.0hz,4h),0.86(t,j=6.8hz,2h).
[0091]
s16.amc-4c的制备
[0092][0093]
在室温下,将中间体21(690mg,0.83mmol)和氢氧化锂即lioh(68mg, 1.66mmol)溶于水(5ml)和thf(10ml)中搅拌反应6h,经液-质联用质谱 仪(即lc-ms)监测反应完全后,将反应液通过高效反相柱制备分离纯化得淡 黄色固体即amc-4c(269mg,收率:39.8%,0.33mmol)。ms(esi)m/z(m+h) +
817.4。1h nmr(400mhz,dmso)δ10.41(s,1h),8.84(s,1h),8.55(d,j=2.4hz, 1h),8.12(d,j=8.8hz,2h),7.96(d,j=9.1hz,1h),7.84(dd,j=9.1,2.3hz,1h), 7.67(d,j=9.0hz,1h),7.59(d,j=8.7hz,2h),7.53(s,1h),5.01(s,1h),3.97(s, 1h),3.60
–
3.52(m,1h),3.29-3.28(m,3h),2.80(s,3h),2.76(s,2h),2.48
–
2.44 (m,2h),2.41(d,j=5.2hz,2h),2.25(s,2h),2.18
–
2.09(m,4h),1.98(s,2h),1.88 (d,j=12.8hz,2h),1.83
–
1.72(m,4h),1.64(s,6h),1.51(s,2h)。
[0094]
2、一种amc-6c半抗原的制备方法,包括如下步骤:
[0095]
s1~s6同上amc-4c半抗原制备过程;
[0096]
s7.中间体23的制备
[0097][0098]
将叔丁氧羰基保护的6-氨基己酸,即化合物22(5.66g,27.8mmol)、hatu (12.7g,
33.4mmol)和diea(10.8g,83.6mmol)溶于dmso(50ml)中并在 室温下继续搅拌反应1小时。将上述混合物溶液缓慢滴加入正在搅拌的中间体9 (10g,41.8mmol)和diea(10.8g,83.6mmol)的dmso(100ml)溶液中, 并在室温下继续搅拌反应半小时,经液-质联用质谱仪(即lc-ms)监测反应完 全后,向反应液中直接加入氯甲酸-9-芴基甲酯即fmoccl(21.63g,83.6mmol) 和diea(10.8g,83.6mmol),并继续搅拌反应1小时,经液-质联用质谱仪(即 lc-ms)监测反应完全后,加入水(300ml),用乙酸乙酯即ea(100ml
×
5) 萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫酸钠干燥过滤后浓缩。 剩余物经柱层析(pe:ea=100:0-35:65)分离纯化得中间体23(8.2g,收率:48.9%, 13.6mmol)的淡黄色固体,即中间体23。ms(esi)m/z(m+h-100)
+
502.1。1h nmr (400mhz,dmso)δ7.88(d,j=7.5hz,2h),7.70(s,2h),7.41(s,2h),7.34(dd,j= 7.4,1.0hz,3h),7.10(s,1h),6.74(t,j=5.3hz,1h),4.18(s,3h),4.03(q,j=7.1 hz,1h),3.00
–
2.79(m,2h),2.11(s,4h),1.90
–
1.70(m,8h),1.36(s,16h),1.18(t, j=7.1hz,4h).
[0099]
s8.中间体24的制备
[0100][0101]
将中间体23(8.2g,13.6mmol)溶于4m的盐酸二氧六环溶液中(50ml) 中并在室温下继续搅拌反应1小时,tlc显示反应完全,将反应液浓缩得淡黄 色油状液体即中间体24(8.12g)。ms(esi)m/z(m+h)
+
502.3。1h nmr(400mhz, dmso)δ7.37(dt,j=32.7,7.4hz,1h),7.37(dt,j=32.7,7.4hz,1h),4.18(s,1h), 3.57(s,2h),2.75
–
2.71(m,0h),2.69(s,1h),2.52
–
2.48(m,1h),2.12(s,1h),2.01 (t,j=7.3hz,0h),1.83(dd,j=36.3,23.0hz,1h),1.50(dtd,j=22.5,15.0,7.5hz, 1h),1.33
–
1.17(m,1h)。
[0102]
s9.中间体25的制备
[0103][0104]
将中间体6(1.8g,5.1mmol)、hatu(2.3g,6.1mmol)和diea(1.9g,15.3 mmol)溶于dcm(20ml)中并在室温下继续搅拌反应1小时。将上述液体加 入正在搅拌的中间体24(2.6g,5.1mmol)的dcm(100ml)溶液中,并在室 温下继续搅拌反应半小时,经液-质联用质谱仪(即lc-ms)监测反应完全后, 加入水(100ml),用dcm(100ml
×
3)萃取,有机层用饱和食盐水洗涤(100 ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经柱层析(pe:ea=100:0-0:100) 分离纯化得橙色固体即15(3.4g,收率:80.4%,4.1mmol)。ms(esi)m/z(m+h) +
837.2。1h nmr(400mhz,dmso)δ8.65(s,1h),7.88(d,j=7.4hz,2h),7.70 (dd,j=8.2,4.0hz,3h),7.40(dd,j=13.7,4.9hz,3h),7.32(td,j=7.4,1.0hz, 3h),7.15(d,j=8.3hz,1h),6.98(s,1h),6.63(dd,j=20.1,2.4hz,2h),6.46(dd,j =8.4,2.4hz,1h),5.76(s,1h),4.27(d,j=5.6hz,2h),4.18(s,3h),3.84(s,3h), 3.73(s,3h),3.28
–
3.21(m,2h),2.69(d,j=1.4hz,4h),2.08(d,j=17.2hz,3h), 1.79(d,j=20.5hz,7h),1.58
–
1.41(m,7h),1.28(dd,j=19.1,12.1hz,7h).
[0105]
s10.中间体26的制备
[0106][0107]
将中间体25(6g,7.17mmol)溶于dcm(100ml)中,加入tfa(10ml) 并在室温下继续搅拌反应5分钟,经液-质联用质谱仪(即lc-ms)监测反应完 全后,将反应液浓缩用氯仿带干3次得16(7g)的红色粘稠液体即为中间体26。 ms(esi)m/z(m+h)
+
687.2。
[0108]
s11.中间体28的制备
[0109][0110]
将己二酸单甲酯,即化合物27(11.5g,71.7mmol)溶于氯仿(100ml)中, 随后加入二氯亚砜(25.7g,215mmol)并在60度下搅拌反应1小时。将上述液 体旋干并用甲苯带干一次后,用dcm(50ml)溶解,随后加入搅拌中的中间 体26(7g)和diea(3.37g,26.2mmol)的dcm(100ml)溶液中,在室温 下搅拌反应半小时,经液-质联用质谱仪(即lc-ms)监测反应完全后,反应液 加水(100ml),水相用ea(100ml
×
3)萃取,有机层用饱和食盐水洗涤(100 ml)后用无水硫酸钠干燥,随后过滤浓缩。剩余物经硅胶柱(pe:ea: meoh=100:0:0-0:100:0-0:0:100)分离纯化得淡黄色固体即中间体28(5g,收率: 84.1%,6.03mmol)。ms(esi)m/z(m+h)
+
829.4。
[0111]
s12.中间体29的制备
[0112][0113]
将中间体28(5g,6.03mmol)于2m的稀盐酸(120ml)和thf(100ml) 中,并在室温下继续搅拌反应12小时,经液-质联用质谱仪(即lc-ms)监测 反应完全后,用ea(150ml
×
3)萃取,有机层用饱和食盐水洗涤(150ml)后 用无水硫酸钠干燥过滤后浓缩得淡黄色固体即中间体29(3g,收率:61.02%, 3.68mmol)。ms(esi)m/z(m+h)
+
815.4。
[0114]
s13.中间体30的制备
[0115][0116]
将中间体29(3g,3.68mmol)、中间体17(1.56g,7.36mmol)、hatu(2.1 g,5.52mmol)和tea(1.86g,18.4mmol)于dmf(100ml),在室温下搅拌过 夜,经液-质联用质谱仪(即lc-ms)监测反应完全后,反应液加水(100ml), 水相用ea(100ml
×
3)萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫 酸钠干燥,随后过滤浓缩。剩余物经反相柱层析(氨水:水:乙腈=100:0-30:70) 分离纯化得淡黄色固体即中间体30(2g,收率:53.8%,1.98mmol)。ms(esi) m/z(m+h)
+
1009.5。
[0117]
s14.中间体31的制备
[0118][0119]
将中间体30(2g,1.98mmol)溶于etoh(20ml)中,加入二乙胺(5ml) 并在室温下继续搅拌反应12小时,经液-质联用质谱仪(即lc-ms)监测反应 完全后,将反应液浓缩,用etoh带干两次。剩余物用水(20ml)溶解,用ea 洗去杂质后冻干得淡黄色固体即中间体31(1.5g,收率:96.2%,1.90mmol)。 ms(esi)m/z(m+h)+787.4。
[0120]
s15.中间体33的制备
[0121][0122]
将中间体31(1.5g,1.9mmol)、4-氧丁酸甲基酯,即化合物20(2g,17.2mmol) 和stab(4g,9.5mmol)于dcm(100ml)和甲醇(10ml)中,在室温下搅 拌过夜,经液-质联用质谱仪(即lc-ms)监测反应完全后,反应液旋干。剩余 物经反相柱(氨水,水:乙腈=100:0-70:30)分离纯化得淡黄色固体即中间体33(1 g,收率:58.9%,1.12mmol)。ms(esi)m/z(m+h)
+
887.4。1h nmr(400mhz, dmso)δ10.35(s,1h),8.81(t,j=5.8hz,1h),8.58(d,j=2.2hz,1h),8.14(d,j= 8.8hz,2h),7.97(d,j=9.1hz,1h),7.86(dd,j=9.1,2.2hz,1h),7.57(t,j=8.3 hz,2h),7.52(d,j=9.2hz,1h),5.82(d,j=4.8hz,1h),5.02(s,1h),4.83(s,1h), 4.17
–
3.96(m,3h),3.58(s,3h),3.17(s,6h),2.78(s,4h),2.40
–
2.20(m,5h),2.01 (dt,j=20.2,11.2hz,8h),1.88
–
1.68(m,6h),1.68
–
1.23(m,23h).
[0123]
s16.amc-6c的制备
[0124][0125]
在室温下,将中间体33(1g,1.12mmol)和lioh(90mg,2.8mmol)溶 于水(10ml)和thf(20ml)中搅拌反应6h,经液-质联用质谱仪(即lc-ms) 监测反应完全后,将反应液通过高效反相柱制备分离纯化得淡黄色固体即 amc-6c(400mg,收率:40.9%,0.458mmol)。ms(esi)m/z(m+h)+873.7。1h nmr(400mhz,dmso)δ10.62(s,1h),8.81(t,j=5.8hz,1h),8.59(d,j=2.0 hz,1h),8.11(d,j=8.8hz,2h),8.04(s,1h),7.93(dt,j=9.1,5.6hz,2h),7.60(d, j=8.7hz,2h),7.34(s,1h),5.76(s,2h),5.03(d,j=2.2hz,1h),4.06
–
3.96(m, 1h),3.58(dd,j=10.3,8.4hz,2h),3.35
–
3.24(m,4h),2.79(s,3h),2.57(s,2h), 2.32(s,2h),2.10(dd,j=13.1,6.7hz,2h),2.02(t,j=6.9hz,7h),1.89
–
1.63(m, 7h),1.63
–
1.24(m,20h)。
[0126]
实施例2多簇串联线性人工抗原的制备
[0127]
本实施例提供多簇串联线性人工抗原的制备方法,主要包括免疫原和包被原 的合成。免疫原与包被原的制备不同之处在于载体种类,所述的免疫原采用的载 体牛乳铁蛋白(lf)、钥孔血蓝蛋白(klh);所述的包被原采用载体蛋白为卵 清蛋白(ova)。免疫原的制备方法。本发明采用的合成免疫原/包被原的方法为 活泼酯法。具体步骤如下:
[0128]
将24.69mg amc-4c/26.38mg amc-6c、8.69mg edc和5.22mg nhs溶于 600μl的dmf中,4℃搅拌反应过夜,记为a液。称取载体蛋白10mg溶于4mlpbs缓冲溶液(0.01m,ph 7.4)
中,搅拌溶解制成b液。磁力搅拌下,吸取a 液逐滴加入到b液中,4℃下磁力搅拌反应12h。将反应液于4℃下用pbs透析 3天,每天换2次透析液。即可得到免疫原,冻存于-20℃冰箱中,备用。取实施 例1制备的半抗原和实施例2制备的人工抗原分别进行紫外波长扫描(150~400 nm)进行鉴定,结果如图1所示,相比于载体蛋白与半抗原,偶联后特征吸收 峰及峰形均发生变化,结果说明抗原偶联成功。
[0129]
实施例3多簇串联线性抗原特异性多克隆抗体的制备
[0130]
动物免疫:用健康的6周龄的balb/c雌鼠作为实验动物,将实施例2制备的 两种免疫原分别与等量的佐剂(第一次用完全弗氏佐剂,之后均用不完全弗氏佐 剂)混合乳化,在小鼠颈背部和腹腔进行皮下注射,每次免疫剂量为0.5ml(其 中含有0.5mg免疫原)。首次免疫用0.5ml完全弗氏佐剂与抗原乳化后用于免 疫,4周后用0.5ml不完全弗氏佐剂与抗原乳化后加强免疫,之后每隔2周免 疫一次,期间尾巴静脉取少量血进行抗体质量鉴定,待抗体稳定后,选择性能最 好的小鼠收集血清,37℃温浴30min,室温离心20min,取上清分装于离心管中, 于-20℃下保存使用。
[0131]
抗血清效果评价:以实施例2制备得到的多簇串联线性包被抗原,取上述采 集的的小鼠血清为检测抗体,采用间接竞争elisa法测定小鼠血清的抗血清效 价与抑制率,综合考虑各抗血清的效价、抑制率,对其进行评价。具体操作步骤 如下:
[0132]
1)包板:将多簇串联线性包被抗原用0.05m碳酸盐缓冲液(ph 9.6)稀释 至1000ng/ml,按100μl/孔,4℃包被过夜;弃包被液,pbst洗涤2次,每孔 加入120μl封闭液(5%脱脂牛奶)37℃封闭3h;弃封闭液,37℃烘干60min, 用密封袋装于4℃待用,得到包好的酶标板。
[0133]
2)血清效价与抑制检测:将步骤1)包好的酶标板,效价列:每孔分别加 入50μl pbs和50μl按梯度倍数稀释的血清;抑制列:每孔加入50μl稀释好 的1000ng/ml药物(多簇串联线性半抗原)和50μl按梯度倍数稀释的血清, 做2组平行。37℃孵育40min,用pbst洗五次,拍干孔内液体,加入1:5000 稀释的酶标二抗(羊抗鼠igg-hrp),37℃孵育30min,用pbst洗五次,拍干 孔内液体,加入100μl tmb底物液,37℃避光显色10min;加入50μl终止液 (10%h2so4)终止反应;用酶标仪读取450nm处的吸光值。
[0134]
3)实验结果
[0135]
基于间接竞争elisa法测定小鼠血清的抗血清效价与抑制率,结果如表1 所示,效价在8~32k之间,对多簇串联线性半抗原的抑制率为63~90%。
[0136]
表1小鼠抗血清效果
[0137][0138]
以上结果表明,本发明制备的多簇串联线性半抗原及其人工抗原具有良好的 免疫原性,可以用于后续制备单克隆抗体、单链抗体和纳米抗体。
[0139]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施 例
的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替 代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1