苯并咪唑衍生物的制作方法
goutieres综合征(ags)。用作ags模型的trex1缺陷型小鼠佐证了cgas/sting的参与。
13.因此,抑制疾病介导细胞因子上游的cgas通路是治疗患者的多种自身免疫性疾病的一种新策略。适应症可以包括与ifn信号传导相关的或由tnfα和il1β驱动的病症。
14.迄今为止,许多由先天性免疫系统的调节异常引起的疾病都缺乏有效的治疗方法。
15.本发明的化合物结合cgas并调节其活性。
16.式(i)化合物特别适用于例如系统性红斑狼疮(sle)、表皮型皮肤病(如皮肌炎或皮肤狼疮)、间质性肺纤维化、干燥综合征、i型糖尿病、炎症性肠病、非酒精性脂肪性肝炎(nash)、青少年炎性关节炎、强直性脊柱炎、痛风或aicardi-goutieres综合征(ags)的治疗或预防。
17.在本说明书中,术语“烷基”在单独或组合下指具有1至8个碳原子的直链或支链烷基、特别是具有1至6个碳原子的直链或支链烷基并且更特别是具有1至4个碳原子的直链或支链烷基。直链和支链c1-c8烷基基团的示例为甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、仲丁基、异构戊基、异构己基、异构庚基和异构辛基,特别地为甲基、乙基、丙基、丁基和戊基。烷基的特定实例是甲基、乙基和丙基。甲基和乙基为式(i)化合物中“烷基”的特定示例。
18.术语“环烷基”在单独或组合下指具有3至8个碳原子的环烷基环、特别是具有3至6个碳原子的环烷基环。环烷基的实例是环丙基、环丁基、环戊基和环己基、环庚基和环辛基。“环烷基”的一个特定实例是环丙基。
19.术语“烷氧基”或“烷基氧基”单独或组合表示式“烷基-o
‑”
的基团,其中术语“烷基”具有先前给出的含义,诸如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基和叔丁氧基。“烷氧基”的一个特定实例为甲氧基。
20.术语“氧基”在单独或组合下指-o-基团。
21.术语“卤素”或“卤代”,单独或组合地,表示氟、氯、溴或碘并且特别地为氟、氯或溴,更特别地为氟或氯。术语“卤代”与另一种基团组合,指经至少一个卤素取代、尤其经一个至五个卤素、特别地一个至四个卤素(即一个、两个、三个或四个卤素)取代的所述基团。
22.术语“卤代烷基”在单独或组合下指经至少一个卤素取代、尤其经一个至五个卤素、特别地一个至三个卤素取代的烷基。特定的“卤代烷基”为氟乙基。
23.术语“羰基”在单独或组合下指-c(o)-基团。
24.术语“氨基”在单独或组合下指伯氨基(-nh2)、仲氨基(-nh-)或叔氨基(-n-)。
25.单独或组合使用的术语“烷基氨基”表示被至少一个烷基取代的氨基基团。具体的“氨基烷基”是氨基甲基、氨基乙基和氨基丙基。优选的“氨基烷基”是氨基乙基和氨基丙基。
26.术语“氨基羰基”、单独或组合下,表示-c(o)nh2基团。
27.术语“磺酰基”单独或组合表示-so
2-基团。
28.术语“药学上可接受的盐”是指那些保留游离碱或游离酸的生物学有效性和特性的盐,其并非在生物学上或其它方面所不希望的。这些盐用无机酸诸如盐酸、氢溴酸、硫酸、硝酸、磷酸(特别是盐酸)和有机酸诸如乙酸、丙酸、乙醇酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸、n-乙酰基半胱氨酸形成。此外,可通过无机碱或有机碱与游离酸的加成制备这些盐。
衍生自无机碱的盐包括但不限于钠盐、钾盐、锂盐、铵盐、钙盐、镁盐。衍生自有机碱的盐包括但不限于与下述有机碱形成的盐:伯胺、仲胺和叔胺,取代胺包括天然出现的取代胺、环状胺和碱性离子交换树脂,诸如异丙胺、三甲胺、二乙胺、三乙胺、三丙胺、乙醇胺、赖氨酸、精氨酸、n-乙基哌啶、哌啶、聚胺树脂。式(i)化合物也可以两性离子的形式存在。式(i)化合物的特别优选的药学上可接受的盐为盐酸、氢溴酸、硫酸、磷酸、乙酸、钠和钾的盐。
29.术语“药用酯”意指通式(i)的化合物可在官能团处衍生化以提供能够在体内转化回母体化合物的衍生物。此类化合物的实例包括生理上可接受的和代谢不稳定的酯衍生物,诸如甲氧基甲酯、甲硫基甲酯和新戊酰氧基甲酯。另外,类似于代谢不稳定的酯,能够在体内产生通式(i)的母体化合物的通式(i)的化合物的任何生理上可接受的等同形式均在本发明的范围内。
30.如果本发明的起始材料或式(i)化合物中的一者含有一个或多个在一个或多个反应步骤的反应条件下不稳定或有反应性的官能团,则可在应用本领域公知方法的关键步骤之前引入适当的保护基团(如例如在t.w.greene和p.g.m.wuts,第3版,1999,wiley,new york的“protective groups in organic chemistry”中所述)。可以使用文献中描述的标准方法,在合成的晚期移除这类保护基。保护基的实例是叔丁氧羰基(boc)、9-芴基甲基氨基甲酸酯(fmoc)、2-三甲基甲硅烷基乙基氨基甲酸酯(teoc)、苄氧基羰基(cbz)和对甲氧基苄氧羰基(moz)。特别优选的保护基团为叔丁氧基羰基(boc)。
31.式(i)的化合物可含有几个不对称中心并且能以光学纯的对映异构体、对映异构体诸如外消旋体的混合物、非对映异构体的混合物、非对映异构外消旋体或非对映异构外消旋体的混合物等形式存在。
32.术语“不对称碳原子”意指具有四个不同取代基的碳原子。依据cahn-ingold-prelog顺序规则,不对称碳原子可为“r”或“s”组态。
33.因此,本发明涉及:
34.根据本发明所述的化合物,其中r1为苯基、甲基羰基氨基丙基、甲基磺酰基氨基丙基、氯苯基甲基、吡啶基甲基、二氨基乙基、甲氧基乙基、环丙基、乙基、三氟乙基、苯甲基或苯乙基;
35.根据本发明所述的化合物,其中r1为苯基烷基;
36.根据本发明所述的化合物,其中r1为苯基甲基或苯基乙基;
37.根据本发明所述的化合物,其中r2为氢;
38.根据本发明所述的化合物,其中r3为卤素;以及
39.根据本发明所述的化合物,其中r3为氯;
40.一种式(i)化合物,其选自
41.5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸;
[0042]
1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸;
[0043]
5-(2-氯-4-甲基苯基)-1-(3-(甲基磺酰氨基)丙基)-1h-苯并[d]咪唑-7-甲酸;
[0044]
5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸;
[0045]
5-(2-氯-4-甲基苯基)-1-(吡啶-4-基甲基)-1h-苯并[d]咪唑-7-甲酸;
[0046]
5-(2-氯-4-甲基苯基)-1-(吡啶-3-基甲基)-1h-苯并[d]咪唑-7-甲酸;
[0047]
6-(2-氯-4-甲基苯基)-3-(吡啶-2-基甲基)苯并咪唑-4-甲酸;
[0048]
5-(2-氯-4-甲基苯基)-1-(2-(二甲基氨基)乙基)-1h-苯并[d]咪唑-7-甲酸;
[0049]
5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸;
[0050]
5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸;
[0051]
5-(2-氯-5-氟-4-甲基苯基)-1-乙基-1h-苯并[d]咪唑-7-甲酸;
[0052]
5-(2-氯-4-甲基苯基)-1-乙基-1h-苯并[d]咪唑-7-甲酸;
[0053]
5-(2-氯-4-甲基苯基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-7-甲酸;
[0054]
5-(2-氯-4-甲基苯基)-1-苯乙基-1h-苯并[d]咪唑-7-甲酸;和
[0055]
1-苄基-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸;
[0056]
或其药学上可接受的盐或酯。
[0057]
一种式(i)化合物,其选自
[0058]
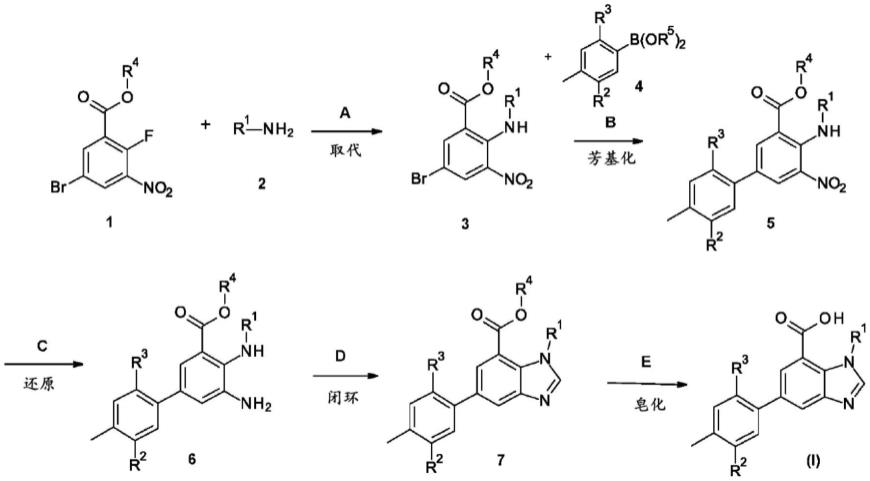
5-(2-氯-4-甲基苯基)-1-苯乙基-1h-苯并[d]咪唑-7-甲酸;和
[0059]
1-苄基-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸;
[0060]
或其药学上可接受的盐或酯。
[0061]
式(i)化合物的合成例如可以根据以下方案来实现。
[0062]
根据本发明的式(i)化合物,其中r1为苯基,其可以根据方案1制备。
[0063]
方案1
[0064][0065]
在方案1中,r1为苯基。r2和r3如上所定义,r4为烷基,r5为氢或烷基。
[0066]
在方案1中,方便地r4为甲基。
[0067]
在方案1中,方便地r5为氢。
[0068]
步骤a:亲核取代可以通过相应的邻氟硝基衍生物1与胺2在20℃至140℃下在合适的溶剂诸如二噁烷、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃、二甲氧基乙烷、二甘醇二甲醚、乙醇或甲醇中,添加或不添加额外的碱如三乙胺、乙基二异丙基胺等,在有或没有微波照射的情况下反应5分钟至18小时来实现。
[0069]
方便的条件是将氟代硝基衍生物与胺在二甲基甲酰胺中在110℃加热1小时。
[0070]
步骤b:溴代衍生物3与合适的硼酸或硼酸酯4的偶联可以通过使用钯催化剂(诸如钯(ii)-乙酸、钯(ii)-氯化物、1,1'-双(二苯基膦基)二茂铁-钯(ii)二氯二氯甲烷复合物、
三(二亚苄基丙酮)二钯、三(二亚苄基丙酮)二钯-氯仿加合物或四(三苯基膦)钯(0)与配体(诸如三苯基膦、三环己基膦、x-phos、xantphos等和碱(诸如磷酸钾、碳酸钾、碳酸铯、三乙胺或二异丙基乙胺)在合适的溶剂(诸如二噁烷、甲苯、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃、二甲氧基乙烷、二甘醇二甲醚、乙醇、甲醇、水)或上述溶剂的混合物中,在有或没有微波辐照的情况下,在20℃至180℃组合使用5分钟至18小时来实现。
[0071]
方便的条件是在二噁烷和水的混合物中在110℃使用三(二亚苄基丙酮)二钯-氯仿加合物、x-phos和磷酸钾2小时。
[0072]
步骤c:硝基基团的还原可以通过使用氢气和催化剂如镍、铂、钯或pd/c(碳载钯5%至10%)在乙酸乙酯、甲醇或乙醇中在大气压或高压下在0℃至70℃来实现。此外,它可以通过各种不同的其他还原剂,诸如使用铁和酸(诸如盐酸的水溶液)或铁和氯化铵水溶液,或氯化锡(ii)的乙醇或乙酸乙酯溶液,或连二亚硫酸钠的水溶液在室温或高温下来实现。
[0073]
方便的条件是在乙酸乙酯中在大气压下在20℃使用氢气和pd/c(10%)18小时。
[0074]
步骤d:形成苯并咪唑7的闭环可以通过使二氨基化合物与甲酸或甲酸衍生物诸如原甲酸三乙酯、原甲酸三甲酯或二甲基甲酰胺二甲基缩醛在室温或升高的温度下,在有或没有额外溶剂的情况下反应来实现。
[0075]
优选的条件是将二氨基化合物在甲酸中加热5分钟。
[0076]
步骤e:皂化可通过烷基酯7与碱诸如氢氧化锂、氢氧化钠、氢氧化钾等,在合适的溶剂诸如水、四氢呋喃、乙醇、甲醇或其混合物中,在0℃至70℃反应1小时至18小时来实现。皂化还可通过使烷基酯7与酸诸如氢溴酸或盐酸在水或乙酸或其混合物中在20℃至110℃下反应1小时至24小时来实现。
[0077]
有利的条件是在四氢呋喃和水的混合物中在65℃使用氢氧化锂18小时。
[0078]
根据本发明的式(i)化合物,其中r1不为苯基,其可以根据方案2制备。
[0079]
方案2
[0080][0081]
在方案2中,r1至r3如上所定义,r4为烷基,r5为氢或烷基。
[0082]
在方案2中,方便地r4为甲基。
[0083]
在方案2中,方便地r5为氢。
[0084]
步骤a:溴代衍生物1与合适的硼酸或硼酸酯2的偶联可以通过在有或没有微波辐照的情况下,将钯催化剂(诸如钯(ii)-乙酸、钯(ii)-氯化物、1,1'-双(二苯基膦基)二茂铁-钯(ii)二氯二氯甲烷复合物、三(二亚苄基丙酮)二钯、三(二亚苄基丙酮)二钯-氯仿加合物或四(三苯基膦)钯(0)与配体(诸如三苯基膦、三环己基膦、x-phos、xantphos等和碱(诸如磷酸钾、碳酸钾、碳酸铯、三乙胺或二异丙基乙胺)在合适的溶剂(诸如二噁烷、甲苯、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃、二甲氧基乙烷、二甘醇二甲醚、乙醇、甲醇、水)或上述溶剂的混合物中在20℃至180℃组合使用5分钟至18小时来实现。
[0085]
方便的条件是在二噁烷和水的混合物中在100℃使用三(二亚苄基丙酮)二钯-氯仿加合物、x-phos和磷酸钾2小时至4小时。
[0086]
步骤b:烷基化/苄基化可以通过使苯并咪唑3与任选取代的卤代烷或任选取代的苄基卤4,诸如取代的烷基氯、取代的烷基溴、取代的烷基碘、取代的烷基、取代的苄基溴、取代的苄基氯等和碱诸如碳酸铯、碳酸钾、碳酸钠、三乙胺或乙基二异丙基胺在溶剂诸如二噁烷、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃中,在0℃至150℃下反应1小时至18小时来实现。如果获得烷基化产物的区域异构混合物,其可以使用有机溶剂诸如庚烷、乙酸乙酯、甲醇和二氯甲烷的混合物通过硅胶柱色谱法进行分离,得到纯化合物的所述区域异构体。
[0087]
烷基化还可以通过苯并咪唑3与合适的硼酸或硼酸酯4(x=b(or5)2)使用铜催化剂诸如乙酸铜(ii)、合适的配体诸如2,2-联吡啶和碱(诸如磷酸钾、碳酸钾、碳酸铯或六甲基二硅氮烷钠)在合适的溶剂(诸如二氯乙烷、二氯甲烷、甲苯、二噁烷、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃或二甲氧基乙烷)中在40℃至120℃下反应数小时来实现。
[0088]
方便的条件是在二甲基甲酰胺中使用取代的烷基溴或苄基溴和碳酸铯,在75℃放置3小时或在室温放置18小时,然后进行硅胶柱色谱法。
[0089]
步骤c:烷基化/苄基化可以通过使苯并咪唑1与任选取代的卤代烷或任选取代的苄基卤4,诸如取代的烷基溴、取代的烷基碘、取代的烷基、取代的苄基溴、取代的苄基氯等和碱诸如碳酸铯、碳酸钾、碳酸钠、三乙胺或乙基二异丙基胺在溶剂诸如二噁烷、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃中,在0℃至150℃下反应1小时至18小时来实现。如果获得烷基化产物的区域异构混合物,其可以使用有机溶剂诸如庚烷、乙酸乙酯、甲醇和二氯甲烷的混合物通过硅胶柱色谱法进行分离,得到纯化合物的所述区域异构体。
[0090]
烷基化还可以通过苯并咪唑1与合适的硼酸或硼酸酯4(x=b(or5)2)使用铜催化剂诸如乙酸铜(ii)、合适的配体诸如2,2-联吡啶和碱(诸如磷酸钾、碳酸钾、碳酸铯或六甲基二硅氮烷钠)在合适的溶剂(诸如二氯乙烷、二氯甲烷、甲苯、二噁烷、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃或二甲氧基乙烷)中在40℃至120℃下反应数小时来实现。
[0091]
方便的条件是在二甲基甲酰胺中使用取代的烷基溴或苄基溴,在75℃放置3小时或在室温放置18小时,然后进行硅胶柱色谱法。
[0092]
步骤d:溴代衍生物5与合适的硼酸或硼酸酯2的偶联可以通过在有或没有微波辐照的情况下,将钯催化剂(诸如钯(ii)-乙酸、钯(ii)-氯化物、1,1'-双(二苯基膦基)二茂铁-钯(ii)二氯二氯甲烷复合物、三(二亚苄基丙酮)二钯、三(二亚苄基丙酮)二钯-氯仿加合物或四(三苯基膦)钯(0)与配体(诸如三苯基膦、三环己基膦、x-phos、xantphos等和碱(诸如磷酸钾、碳酸钾、碳酸铯、三乙胺或二异丙基乙胺)在合适的溶剂(诸如二噁烷、甲苯、二甲基乙酰胺、二甲基甲酰胺、四氢呋喃、二甲氧基乙烷、二甘醇二甲醚、乙醇、甲醇、水)或上述溶剂的混合物中在20℃至180℃组合使用5分钟至18小时来实现。
[0093]
方便的条件是在二噁烷和水的混合物中在100℃使用三(二亚苄基丙酮)二钯-氯仿加合物、x-phos和磷酸铯2小时至4小时。
[0094]
步骤e:皂化可通过烷基酯6与碱诸如氢氧化锂、氢氧化钠、氢氧化钾等,在合适的溶剂诸如水、四氢呋喃、乙醇、甲醇或其混合物中,在0℃至70℃反应1小时至18小时来实现。皂化还可通过使烷基酯7与酸诸如氢溴酸或盐酸在水或乙酸或其混合物中在20℃至110℃下反应1小时至24小时来实现。
[0095]
有利的条件是在四氢呋喃和水的混合物中在65℃使用氢氧化锂18小时。
[0096]
因此,本发明还涉及用于制备根据本发明的化合物的方法,该方法包括使式(a1)化合物
[0097]
[0098]
在碱或酸存在下发生皂化;
[0099]
其中r1、r2和r3如上所定义,并且r4为烷基。
[0100]
r4方便地为甲基。
[0101]
皂化的溶剂方便地为水、四氢呋喃、乙醇、甲醇、乙酸或其混合物。
[0102]
在皂化中,碱可以是例如氢氧化锂、氢氧化钠或氢氧化钾。
[0103]
在皂化中,酸可以是例如氢溴酸或盐酸。
[0104]
用于皂化的方便的条件是0℃至110℃放置1小时至24小时。
[0105]
在碱性条件下皂化的优选条件是在四氢呋喃和水的混合物中在65℃使用氢氧化锂18小时。
[0106]
在酸性条件下皂化的优选条件是在乙酸中在110℃使用氢溴酸18小时。
[0107]
本发明还涉及根据本发明的方法制备的根据本发明所述的化合物。
[0108]
本发明的另一实施例提供含有本发明的化合物和治疗惰性载体、稀释剂或赋形剂的药物组合物或药物,以及使用本发明的化合物制备该组合物和药物的方法。在一个示例中,式(i)化合物可以通过在环境温度在适当的ph和期望的纯度下与生理学上可接受的载体(即在所用剂量和浓度下对接受者无毒的载体)混合而配制为盖伦(galenical)施用形式。制剂的ph主要取决于化合物的具体用途和浓度,但是优选在约3至约8的范围内。在一个示例中,将式(i)化合物在ph 5的乙酸盐缓冲液中配制。在另一实施例中,式(i)化合物是无菌的。化合物可以例如作为固体或无定形组合物、作为冻干制剂或作为水溶液储存。
[0109]
以与良好医学实践一致的方式配制、计量和施用组合物。在这种情况下需要考虑的因素包括所治疗的特定疾患、所治疗的特定哺乳动物、个体患者的临床病症、疾患的原因、药剂的递送部位、施用方法、施用的时间安排,以及执业医师已知的其他因素。
[0110]
本发明的化合物可通过任何适合的方式施用,包括口服、局部(包括颊和舌下)、直肠、阴道、经皮、肠胃外、皮下、腹膜内、肺内、皮内、鞘内和硬膜外和鼻内,以及(如果需要用于局部治疗)病灶内施用。肠胃外输注包括肌内、静脉内、动脉内、腹膜内或皮下施用。
[0111]
本发明化合物可以任何方便的施用形式施用,例如,片剂、粉剂、胶囊剂、溶液剂、分散剂、混悬剂、糖浆剂、喷雾剂、栓剂、凝胶剂、乳剂、贴剂等。此类组合物可以包含药物制剂中常规的组分,例如,稀释剂、载体、ph调节剂、甜味剂、填充剂和其他活性剂。
[0112]
通过混合本发明的化合物和载体或赋形剂来制备通常的制剂。适合的载体和赋形剂是本领域技术人员熟知的,并且在例如ansel,howard c.等人,ansel’s pharmaceutical dosage forms and drug delivery systems.philadelphia:lippincott,williams和wilkins,2004;gennaro,alfonso r.等人remington:the science and practice of pharmacy.philadelphia:lippincott,williams&wilkins,2000;以及rowe,raymond c.handbook of pharmaceutical excipients.chicago,pharmaceutical press,2005中有详细描述。制剂还可以包含一种或多种缓冲剂、稳定剂、表面活性剂、湿润剂、润滑剂、乳化剂、悬浮剂、防腐剂、抗氧化剂、不透明剂、助流剂、加工助剂、着色剂、甜味剂、加香剂、调味剂、稀释剂和其他已知的添加剂,以提供美观的药物(例如,本发明的化合物或其药物组合物)展示或有助于药物产品(例如,药物)的制备。
[0113]
本发明还特别涉及:
[0114]
式(i)化合物用于系统性红斑狼疮(sle)、表皮型皮肤病(如皮肌炎或皮肤狼疮)、
间质性肺纤维化、干燥综合征、i型糖尿病、炎症性肠病、非酒精性脂肪性肝炎(nash)、青少年炎性关节炎、强直性脊柱炎、痛风或aicardi-goutieres综合征(ags)的治疗或预防的用途;
[0115]
式(i)化合物用于制备用于系统性红斑狼疮(sle)、表皮型皮肤病(如皮肌炎或皮肤狼疮)、间质性肺纤维化、干燥综合征、i型糖尿病、炎症性肠病、非酒精性脂肪性肝炎(nash)、青少年炎性关节炎、强直性脊柱炎、痛风或aicardi-goutieres综合征(ags)的治疗或预防的药物的用途;
[0116]
一种用于系统性红斑狼疮(sle)、表皮型皮肤病(如皮肌炎或皮肤狼疮)、间质性肺纤维化、干燥综合征、i型糖尿病、炎症性肠病、非酒精性脂肪性肝炎(nash)、青少年炎性关节炎、强直性脊柱炎、痛风或aicardi-goutieres综合征(ags)的治疗或预防的式(i)化合物;以及
[0117]
一种用于系统性红斑狼疮(sle)、表皮型皮肤病(如皮肌炎或皮肤狼疮)、间质性肺纤维化、干燥综合征、i型糖尿病、炎症性肠病、非酒精性脂肪性肝炎(nash)、青少年炎性关节炎、强直性脊柱炎、痛风或aicardi-goutieres综合征(ags)的治疗或预防的方法,该方法包括向有需要的患者施用有效量的式(i)化合物。
[0118]
现在将通过以下实例说明本发明,所述实例不具有限制性。
[0119]
实例
[0120]
缩写
[0121]
dcm=二氯甲烷;dipea=二异丙基乙胺;dmso=二甲亚砜;esi=电喷雾电离;etoac=乙酸乙酯;meoh=甲醇;ms=质谱;rt=室温;thf=四氢呋喃。
[0122]
实例1
[0123]
5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸
[0124][0125]
a)5-溴-3-硝基-2-(苯基氨基)苯甲酸甲酯
[0126]
在25ml小瓶中,5-溴-2-氟-3-硝基苯甲酸甲酯(1g,3.6mmol,eq:1)与二甲基甲酰胺(10ml)混合。加入苯胺(1.07g,1.05ml,11.5mmol,eq:3.2),将小瓶封闭并加热至110℃保持1h。将粗反应混合物在真空中浓缩,倒入100ml 2m hcl中,并用etoac(3x 150ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,40g,0%至40%的etoac的庚烷溶液)纯化粗物质,以提供红色固体状的标题化合物5-溴-3-硝基-2-(苯基氨基)苯甲酸甲酯(1.11g,3.13mmol,产率87.1%),ms(esi):352.96[m+h]+。
[0127]
b)2'-氯-4'-甲基-5-硝基-4-(苯氨基)-[1,1'-联苯]-3-甲酸甲酯
[0128]
向5-溴-3-硝基-2-(苯基氨基)苯甲酸甲酯(500mg,1.42mmol,eq:1)的二噁烷(8ml)溶液中加入(2-氯-4-甲基苯基)硼酸(243mg,1.42mmol,eq:1)。加入溶解在水(2ml)中
的磷酸钾(604mg,2.85mmol,eq:2)。混合物在x-phos(33.9mg,71.2μmol,eq:0.05)之前2分钟期间用氩气脱气,并加入三(二亚苄基丙酮)二钯-氯仿加合物(36.8mg,35.6μmol,eq:0.025)。将混合物加热至110℃保持2小时。将反应混合物倒入100ml的水中,并用etoac(3x 100ml)萃取。有机层经mgso4干燥并在真空中浓缩。通过色谱法(硅胶,80g,0%至30%的etoac的庚烷溶液)纯化粗物质,以提供红色固体状的标题化合物2'-氯-4-甲基-5-硝基-4-(苯基氨基)-[1,1'-联苯]-3-甲酸甲酯(520mg,966μmol,产率67.9%),ms(esi):397.14[m+h]+。
[0129]
c)氨基-2'-氯-4'-甲基-4-(苯基氨基)-[1,1'-联苯]-3-甲酸盐
[0130]
在25ml圆底烧瓶中,将2'-氯-4'-甲基-5-硝基-4-(苯基氨基)-[1,1'-联苯]-3-甲酸甲酯(450mg,1.13mmol,eq:1)与乙酸乙酯(9ml)混合。在加入pd-c(54.3mg,51μmol,eq:0.045)之前,用氩气替换空气数次,然后用h2替换氩气数次。将反应物在室温搅拌过夜。为进行后处理,用氩气交换h2并将反应物过滤。浓缩溶液并将残余物在真空中干燥。通过色谱法(硅胶,12g,0%至50%的etoac的庚烷溶液)纯化粗物质,以提供淡黄色固体状的标题化合物5-氨基-2'-氯-4'-甲基-4-(苯基氨基)-[1,1'-联苯]-3-甲酸甲酯(306mg,763μmol,产率67.2%),ms(esi):367.18[m+h]+。d)5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸甲酯
[0131]
在10ml圆底烧瓶中,将5-氨基-2'-氯-4'-甲基-4-(苯基氨基)-[1,1'-联苯]-3-甲酸甲酯(50mg,136μmol,eq:1)和甲酸(627mg,523μl,13.6mmol,eq:100)合并。将反应混合物加热回流5分钟,然后缓慢倒入20ml饱和nahco3溶液中并用etoac(3
×
25ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至50%的etoac的庚烷溶液)纯化粗物质,以得到白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸甲酯(34mg,83μmol,产率60.9%),ms(esi):377.15[m+h]+。
[0132]
e)5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸
[0133]
向5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸甲酯(26.6mg,70.6μmol,eq:1)的thf(700μl)溶液中加入溶解在水(350μl)中的lioh(5.92mg,141μmol,eq:2)。将反应混合物加热至65℃并搅拌,直至反应完成。将反应物用hcl(70.6μl,141μmol,eq:2)萃取,加入15ml的水并且将混合物用etoac(3x 25ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至100%的etoac的庚烷溶液,然后0%至7%的meoh的dcm溶液)纯化粗物质,以提供白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-苯基-1h-苯并[d]咪唑-7-甲酸(15mg,38.8μmol,产率55%),ms(esi):363.16[m+h]+。
[0134]
实例2
[0135]
1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸
[0136][0137]
a)6-溴-1h-苯并[d]咪唑-4-甲酸甲酯
[0138]
在250ml圆底烧瓶中,将6-溴-1h-苯并[d]咪唑-4-甲酸(2.9g,12mmol,eq:1)与甲醇(100ml)合并,以得到浅棕色悬浮液。在0℃加入硫酸(11.8g,6.41ml,120mmol,eq:10)萃取,然后将反应混合物加热至65℃并且搅拌过夜。将粗反应混合物在真空中浓缩。将反应混合物倒入100ml饱和nahco3溶液中并用etoac(3x 150ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥,以得到灰白色固体状的标题化合物6-溴-1h-苯并[d]咪唑-4-甲酸甲酯(1.97g,7.63mmol,产率63.5%),ms(esi):255.04,257.05[m+h]+。
[0139]
b)6-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-4-甲酸甲酯
[0140]
在100ml圆底烧瓶中,将6-溴-1h-苯并[d]咪唑-4-甲酸甲酯(2g,7.84mmol,eq:1)、(2-氯-4-甲基苯基)硼酸(1.34g,7.84mmol,eq:1)和磷酸(三)钾(3.33g,15.7mmol,eq:2)与二噁烷(40ml)和水(10ml)混合。在氩气下,加入三(二亚苄基丙酮)二钯-氯仿加合物(203mg,196μmol,eq:0.025)和x-phos(187mg,392μmol,eq:0.05)。将反应加热至100℃并搅拌4小时。将反应混合物倒入50ml饱和nahco3溶液中并用etoac(3x 100ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,80g,0%至80%的etoac的庚烷溶液)纯化粗物质,以提供白色固体状的标题化合物6-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-4-甲酸甲酯(852mg,2.69mmol,产率68.6%),ms(esi):301.12[m+h]+。
[0141]
c)1-(3-((叔丁氧基羰基)氨基)丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯
[0142]
在50ml圆底烧瓶中,将6-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-4-甲酸甲酯(780mg,2.46mmol,eq:1)和碳酸铯(1.27g,3.89mmol,eq:1.58)的混合物与二甲基甲酰胺(20ml)混合,加入(3-溴丙基)氨基甲酸叔丁酯(927mg,3.89mmol,eq:1.58),并且将反应物在室温搅拌过夜。将反应混合物倒入150ml的饱和nacl溶液中,并用etoac(3x 200ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,40g,0%至100%的etoac的庚烷溶液)纯化粗物质,以提供灰白色固体状的标题化合物1-(3-((叔丁氧基羰基)氨基)丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯(571mg,1.25mmol,产率50.6%),ms(esi):458.185[m+h]+。
[0143]
d)1-(3-氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯盐酸盐
[0144]
在25ml圆底烧瓶中,将1-(3-((叔丁氧基羰基)氨基)丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯(571mg,1.25mmol,eq:1)和hcl的二噁烷(1.56ml,
6.23mmol,eq:5)溶液与二噁烷(8ml)混合。将反应物在室温搅拌过夜。将反应物在真空中浓缩,以提供在下一步骤中使用的白色泡沫状的标题化合物1-(3-氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯盐酸盐(315mg,880μmol,产率70.6%)。
[0145]
e)1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯
[0146]
在5ml圆底烧瓶中,将1-(3-氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯(60mg,168μmol,eq:1)和dipea(65mg,87.9μl,503μmol,eq:3)与二氯甲烷(2ml)混合。在0℃加入乙酰氯(14.5mg,13.1μl,184μmol,eq:1.1)。将反应物在室温搅拌1小时。将反应混合物倒入20ml饱和nahco3溶液中并用etoac(3x 25ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至10%的meoh的dcm烷溶液)纯化粗物质,以提供白色固体状的标题化合物1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯(17.7mg,42.2μmol,产率25.2%),ms(esi):400.25[m+h]+。
[0147]
f)1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸
[0148]
向1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸甲酯(17.7mg,44.3μmol,eq:1)的四氢呋喃(500μl)的淡黄色溶液中,加入溶解在水(250μl)中的一水合氢氧化锂(3.71mg,88.5μmol,eq:2)。将混合物加热至65℃并搅拌过夜。将混合物在真空中浓缩。残余物用甲基四氢呋喃(2ml)吸收,形成的晶体通过sartorius漏斗过滤,以得到灰白色固体状的标题化合物1-(3-乙酰氨基丙基)-5-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-7-甲酸(17.9mg,44.3μmol,产率100%),ms(esi):386.14[m+h]+。
[0149]
实例3
[0150]
5-(2-氯-4-甲基苯基)-1-(3-(甲基磺酰胺基)丙基)-1h-苯并[d]咪唑-7-甲酸
[0151][0152]
使用甲基磺酰氯代替步骤e)中的乙酰氯,以类似于实例2所述程序的相当产率获得白色固体状的标题化合物,ms(esi):422.21[m+h]+。
[0153]
实例4
[0154]
5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸
[0155][0156]
a)5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸甲酯
[0157]
在25ml圆底烧瓶中,将6-(2-氯-4-甲基苯基)-1h-苯并[d]咪唑-4-甲酸甲酯(参见实例2,150mg,474μmol,eq:1)和碳酸铯(247mg,758μmol,eq:1.6)与二甲基甲酰胺(3.5ml)混合。加入3-氯苄基溴(97.4mg,62.1μl,474μmol,eq:1)并将反应物在75℃搅拌3小时。将反应混合物倒入20ml饱和nacl溶液中,并用etoac(3x 50ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至10%的meoh的dcm烷溶液)纯化粗物质,以提供标题化合物5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸甲酯(102mg,240μmol,产率50.6%),ms(esi):425.2[m+h]+。
[0158]
b)5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸
[0159]
在5ml小瓶中,将5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸甲酯(95mg,223μmol,eq:1)和lioh一水合物(14.1mg,335μmol,eq:1.5)与thf(1.2ml)和水(600μl)混合。将反应混合物在室温搅拌3小时。为进行后处理,加入hcl(1m,335μl,335μmol,eq:1.5)。将反应混合物倒入20ml的水中,并用etoac(3x 25ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥,以得到白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-(3-氯苄基)-1h-苯并[d]咪唑-7-甲酸(86.6mg,204μmol,产率91.5%),ms(esi):411.19[m+h]+。
[0160]
实例5
[0161]
5-(2-氯-4-甲基苯基)-1-(吡啶-4-基甲基)-1h-苯并[d]咪唑-7-甲酸
[0162][0163]
使用4-(氯甲基)吡啶盐酸盐代替步骤a)中的3-氯苄基溴,以类似于实例4所述程序的相当产率获得白色固体状的标题化合物,ms(esi):378.17[m+h]+。
[0164]
实例6
[0165]
5-(2-氯-4-甲基苯基)-1-(吡啶-3-基甲基)-1h-苯并[d]咪唑-7-甲酸
[0166][0167]
使用3-(氯甲基)吡啶盐酸盐代替步骤a)中的3-氯苄基溴,以类似于实例4所述程序的相当产率获得白色固体状的标题化合物,ms(esi):378.17[m+h]+。
[0168]
实例7
[0169]
6-(2-氯-4-甲基苯基)-3-(吡啶-2-基甲基)苯并咪唑-4-甲酸
[0170][0171]
使用2-(氯甲基)吡啶盐酸盐代替步骤a)中的3-氯苄基溴,以类似于实例4所述程序的相当产率获得灰白色固体状的标题化合物,ms(esi):378.100[m+h]+。
[0172]
实例8
[0173]
5-(2-氯-4-甲基苯基)-1-(2-(二甲基氨基)乙基)-1h-苯并[d]咪唑-7-甲酸
[0174][0175]
使用2-溴-n,n-二甲基乙-1-胺氢溴酸盐代替步骤a)中的3-氯苄基溴,以类似于实例4所述程序的相当产率获得白色固体状的标题化合物,ms(esi):358.23[m+h]+。
[0176]
实例9
[0177]
5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸
[0178][0179]
a)5-溴-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯
[0180]
在25ml圆底烧瓶中,将6-溴-1h-苯并[d]咪唑-4-甲酸甲酯(250mg,903μmol,eq:1)和碳酸铯(465mg,1.43mmol,eq:1.58)与二甲基甲酰胺(8ml)混合。加入1-溴-2-甲氧基乙烷(198mg,134μl,1.43mmol,eq:1.58),并且将反应物在室温搅拌过夜。将反应混合物倒入50ml饱和nacl溶液中,并用etoac(3x 75ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至80%的etoac的庚烷溶液)纯化粗物质,以提供白色固体状的标题化合物5-溴-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯(142.6mg,455μmol,产率50.4%),ms(esi):313.0,315.0[m+h]+。
[0181]
b)5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯
[0182]
在5ml小瓶中,将5-溴-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯(50mg,160μmol,eq:1)、(2-氯-4-甲基苯基)硼酸(27.2mg,160μmol,eq:1)和磷酸(三)钾(67.8mg,319μmol,eq:2)与二噁烷(1ml)和水(250μl)混合。小瓶在x-phos(3.81mg,7.98μmol,eq:0.05)之前用氩气脱气,并加入三(二亚苄基丙酮)二钯-氯仿加合物(4.13mg,3.99μmol,eq:0.025)。将小瓶封闭并将反应混合物加热至110℃并搅拌2小时。为进行后处理,将反应混合物倒入20ml水中并用etoac(3
×
25ml)萃取。合并有机层,经mgso4干燥,通过烧结玻璃过滤,在真空中浓缩并干燥。通过色谱法(硅胶,12g,0%至5%的meoh的dcm烷溶液)纯化粗物质,以提供灰白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯(41.5mg,107μmol,产率66.9%),ms(esi):359.21[m+h]+。
[0183]
c)5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸
[0184]
在10ml圆底烧瓶中,将5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸甲酯(38mg,106μmol,eq:1)和lioh一水合物(6.67mg,159μmol,eq:1.5)与thf(0.75ml)和水(375μl)混合。将反应混合物加热至65℃并搅拌3小时。用hcl(0.1m,159μl,159μmol,eq:1.5)将反应猝灭,倒入20ml的水中,并用etoac(3x 25ml)萃取。合并有机层,在真空中浓缩并干燥,以得到白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-(2-甲氧基乙基)-1h-苯并[d]咪唑-7-甲酸(35.9mg,96.6μmol,产率91.2%),ms(esi):345.12[m+h]+。
[0185]
实例10
[0186]
5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸
[0187][0188]
a)5-溴-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯
[0189]
将6-溴-1h-苯并[d]咪唑-4-甲酸甲酯(302mg,1.18mmol,eq:1)、环丙基硼酸(265mg,2.96mmol,eq:2.5)和碳酸钠(314mg,2.96mmol,eq:2.5)悬浮在1,2-二氯乙烷(15ml)中。在室温下,乙酸铜(ii)溶液(263mg,1.42mmol,eq:1.2)和2,2-联吡啶(224mg,1.42mmol,eq:1.2)的1,2-二氯乙烷(20ml,在70℃制备)溶液中滴加。将反应混合物在80℃(油浴温度)搅拌过夜。为进行后处理,将反应混合物用二氯甲烷稀释并依次用饱和nh4cl溶液和饱和nacl溶液洗涤。有机层用na2so4干燥,并且在真空中浓缩。将残余物通过柱色谱法(硅胶,0%至100%的etoac的庚烷溶液,然后10% meoh的ch2cl2溶液)纯化,以产出黄色固体状的标题化合物5-溴-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯(16.4mg,55.5μmol,产率4.7%),ms(esi):295.0,297.0[m+h]+。
[0190]
b)5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯
[0191]
将5-溴-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯(14.7mg,49.8μmol,eq:1)和(2-氯-4-甲基苯基)硼酸(12.7mg,74.7μmol,eq:1.5)溶解于1,4-二噁烷(800μl)和水(400μl)中。加入碳酸铯(65.6mg,199μmol,eq:4)并通过向混合物中通入氩气(5分钟)使混合物脱气,然后加入1,1'-双(二苯基膦基)二茂铁-二氯化钯(ii)二氯甲烷络合物(2.03mg,2.49μmol,eq:0.05)。将反应在密封管中在90℃(油浴温度)搅拌15分钟。为进行后处理,将混合物在etoac中吸收并依次用饱和nh4cl溶液和盐水洗涤,有机层经na2so4干燥,过滤并蒸发。将残余物通过柱色谱法(硅胶,0%至100%的etoac的庚烷溶液)纯化,以产出无色油状的标题化合物5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯(12mg,35.2μmol,产率70.7%),ms(esi):341.1[m+h]+。
[0192]
c)5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸
[0193]
将5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸甲酯(12mg,35.2μmol,eq:1)溶于thf(500μl)中。在室温加入lioh溶液(1m,105.6μl,105.6μmol,eq:3)。将混合物在65℃搅拌2天。为进行后处理,将反应混合物用水稀释并用乙醚萃取两次,有机层用水洗涤,且合并的水层用hcl(2m,79.2μl,158μmol,eq:4.5)并调节ph至3。用2-甲基四氢呋喃萃取两次,且合并的有机层经na2so4干燥,过滤并蒸发,以提供白色固体状的标题化合物5-(2-氯-4-甲基苯基)-1-环丙基-1h-苯并[d]咪唑-7-甲酸(10mg,30.6μmol,产率86.9%),ms(esi):327.1[m+h]+。
[0194]
实例11
[0195]
5-(2-氯-5-氟-4-甲基苯基)-1-乙基-1h-苯并[d]咪唑-7-甲酸
[0196][0197]
使用碘乙烷代替步骤a)中的1-溴-2-甲氧基乙烷和(2-氯-5-氟-4-甲基苯基)硼酸代替步骤b)中的(2-氯-4-甲基苯基)硼酸,以类似于实例9所述程序的相当产率获得白色固体状的标题化合物,ms(esi):333.1[m+h]+。
[0198]
实例12
[0199]
5-(2-氯-4-甲基苯基)-1-乙基-1h-苯并[d]咪唑-7-甲酸
[0200][0201]
使用碘乙烷代替步骤a)中的1-溴-2-甲氧基乙烷,以类似于实例9所述程序的相当产率获得白色固体状的标题化合物,ms(esi):315.1[m+h]+。
[0202]
实例13
[0203]
5-(2-氯-4-甲基苯基)-1-(2,2,2-三氟乙基)-1h-苯并[d]咪唑-7-甲酸
[0204][0205]
使用三氟甲磺酸2,2,2-三氟乙酯代替步骤a)中的1-溴-2-甲氧基乙烷和碳酸钾代替步骤a)中的碳酸铯,以类似于实例9所述程序的相当产率获得白色固体状的标题化合物,ms(esi):369.1[m+h]+。
[0206]
实例14
[0207]
5-(2-氯-4-甲基苯基)-1-苯乙基-1h-苯并[d]咪唑-7-甲酸
reader(perkin elmer)上收集620nm的吸光度数据,并使用以下测量设置:激发滤光片光度计为620nm;从顶部激发;测量高度为1mm;闪光次数为30;集成的闪光灯数量为1。
[0218]
检查所有板是否异常,使用3*sd规则排除空白对照(无蛋白质,第1行)和中性对照(无化合物,第2行)中的异常值。通过空白对照和中性对照将数据归一化为0和100%,并使用4参数逻辑斯谛方程对每条曲线进行拟合和判断以确定cgas抑制作用的ic50。
[0219]
该测定的结果在表1中提供。表1提供了针对本发明的特定示例获得的、如通过上述测定测量的cgas抑制作用的ic50值(μm)。
[0220][0221]
实例a
[0222]
含有以下成分的膜包衣片剂能以常规方式制备:
[0223]
成分每个片剂 内核:
ꢀꢀ
式(i)化合物10.0mg200.0mg微晶纤维素23.5mg43.5mg含水乳糖60.0mg70.0mg聚维酮k3012.5mg15.0mg羟基乙酸淀粉钠12.5mg17.0mg硬脂酸镁1.5mg4.5mg(内核重量)120.0mg350.0mg膜包衣:
ꢀꢀ
羟丙基甲基纤维素3.5mg7.0mg聚乙二醇60000.8mg1.6mg滑石1.3mg2.6mg氧化铁(黄色)0.8mg1.6mg二氧化钛0.8mg1.6mg
[0224]
将活性成分过筛并与微晶纤维素混合,并将混合物用聚乙烯吡咯烷酮的水溶液制粒。然后将颗粒与羟基乙酸淀粉钠和硬脂酸镁混合并压制以得到分别为120或350mg的内核。用上述膜包衣的水溶液/悬浮液对内核上漆。
[0225]
实例b
[0226]
含有以下成分的胶囊能以常规方式制备:
[0227]
成分每个胶囊式(i)化合物25.0mg乳糖150.0mg玉米淀粉20.0mg滑石5.0mg
[0228]
将组分过筛并混合,并填充到大小为2的胶囊中。
[0229]
实例c
[0230]
注射溶液可以具有以下成分:
[0231]
式(i)化合物3.0mg聚乙二醇400150.0mg醋酸适量,将ph调节至5.0注射用水加至1.0ml
[0232]
在聚乙二醇400和注射用水(部分)的混合物中溶解活性成分。通过添加乙酸将ph调节至5.0。通过加入余量的水将体积调节至1.0ml。过滤溶液,使用适宜的超量填充到小瓶中并灭菌。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1