聚烯烃系树脂发泡片及叠层体的制作方法
1.本发明涉及具有优异的柔软性和成型性的聚烯烃系树脂发泡片、以及叠层体。
背景技术:
2.一直以来,以聚烯烃系树脂作为基材树脂的交联发泡片由于柔软性、耐热性和机械强度等优异,因此例如,作为顶蓬、车门面板、仪表板等汽车内装材料而被使用。在这些用途中,以通过适度的柔软性而赋予高级感、在与人接触的扶手部等中赋予减轻负担的功能性等作为目的,提高了柔软性的发泡体的需求增加。
3.作为这样的聚烯烃系树脂发泡片,提出了一种聚烯烃系树脂发泡片,其特征在于,包含熔点为115℃以上且熔体指数为0.1g/10分钟以上且40g/10分钟以下(190℃)的烯烃系嵌段共聚物15质量份以上且75质量份以下,包含熔体指数为0.1g/10分钟以上且25g/10分钟以下(230℃)的聚丙烯系树脂25质量份以上且85质量份以下,凝胶分率为20%以上且75%以下,密度为25kg/m3以上且250kg/m3以下(例如,参照专利文献1)。
4.进一步,提出了一种使用聚烯烃树脂发泡体而成的叠层体以及汽车内装材料,上述叠层体的特征在于,是聚烯烃系树脂发泡体与表皮体的叠层体,聚烯烃系树脂发泡体在构成该聚烯烃树脂发泡体的聚烯烃系树脂100质量%中,包含聚丙烯系树脂30质量%以上且60质量%以下、聚乙烯系树脂1质量%以上且20质量%以下、热塑性弹性体树脂30质量%以上(例如,参照专利文献2)。
5.上述聚烯烃系树脂发泡片和聚烯烃系树脂发泡体的制造方法没有特别限制,但可以大致分为将树脂组合物成型为片状而获得发泡性片的工序、将上述发泡性片交联的工序、将交联了的发泡性片加热发泡而获得发泡片的工序。考虑生产性,在获得发泡片的工序中,有很多将卷状的交联了的发泡性片连续地供给到热媒而发泡,作为卷状的发泡片而卷绕。此时,虽然与发泡的程度有关,但将卷绕速度除以开卷速度而得的md方向拉伸倍率一般在超过3.0的条件下被实施。为了防止发泡时的松弛、褶皱,提升发泡时的md方向拉伸倍率时生产效率好,特别是,在含有聚烯烃系弹性体树脂的情况下,具有粘贴至辊等的的担心等,因此在拉伸倍率高的状态下被生产。
6.现有技术文献
7.专利文献
8.专利文献1:日本特开2015-187232号公报
9.专利文献2:日本特开2016-155344号公报
技术实现要素:
10.发明所要解决的课题
11.专利文献1和2所公开的、聚烯烃系树脂发泡片和使用了聚烯烃系树脂发泡体的叠层体虽然具有优异的柔软性,但与由成型加工时的加热收缩引起的尺寸缺损、由褶皱引起的外观不良等成型性有关的研究不充分,具有成型性不充分这样的问题。
12.因此,本发明的目的是提供具有优异的柔软性和成型性的、聚烯烃系树脂发泡片及其叠层体。
13.用于解决课题的方法
14.本发明人等为了达到上述课题而反复进行了深入研究,结果发现,下述聚烯烃系树脂发泡片具备优异的柔软性和成型性,该聚烯烃系树脂发泡片将包含聚乙烯系树脂0质量%以上且30质量%以下、聚丙烯系树脂30质量%以上且80质量%以下和聚烯烃系弹性体20质量%以上且40质量%以下的树脂混合物作为基材树脂,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的加热尺寸变化率为-35%以上且0%以下。
15.此外,发现下述聚烯烃系树脂发泡片也具备优异的柔软性和成型性,该聚烯烃系树脂发泡片将25%压缩应力(kpa)除以密度(kg/m3)而得的值为2.5以下,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的加热尺寸变化率为-35%以上且0%以下,从而完成了本发明。
16.本发明涉及下述(1)~(12)。
17.(1)一种聚烯烃系树脂发泡片,其将包含聚乙烯系树脂0质量%以上且30质量%以下、聚丙烯系树脂30质量%以上且80质量%以下和聚烯烃系弹性体20质量%以上且40质量%以下的树脂混合物作为基材树脂,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的md方向和td方向的加热尺寸变化率为-35%以上且0%以下。
18.(2)一种聚烯烃系树脂发泡片,其将25%压缩应力(kpa)除以密度(kg/m3)而得的值为2.5以下,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的md方向和td方向的加热尺寸变化率为-35%以上且0%以下。
19.(3)根据(1)或(2)所述的聚烯烃系树脂发泡片,其厚度为1mm以上且5mm以下,密度为40kg/m3以上且100kg/m3以下,凝胶分率为30%以上且60%以下。
20.(4)根据(1)~(3)中任一项所述的聚烯烃系树脂发泡片,其在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下,加热了10分钟时的加热尺寸变化率的md方向/td方向比为0.5以上且1.5以下。
21.(5)根据(1)~(4)中任一项所述的聚烯烃系树脂发泡片,其在比在dsc测定中作为最高熔融峰的最高熔点低20℃的温度下加热了10分钟时的md方向和td方向的加热尺寸变化率为-5%以上且0%以下。
22.(6)根据(1)~(5)中任一项所述的聚烯烃系树脂发泡片,其将md方向的平均气泡直径bd
md
除以td方向的平均气泡直径bd
td
而得的平均气泡直径比bd
md
/bd
td
为0.7以上且1.3以下。
23.(7)根据(1)~(6)中任一项所述的聚烯烃系树脂发泡片,其23℃下的抗拉强度的md方向/td方向比为0.7以上且1.3以下。
24.(8)根据(1)~(7)中任一项所述的聚烯烃系树脂发泡片,其在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的卷曲高度为发泡片厚度以上且15mm以下。
25.(9)根据(1)~(8)中任一项所述的聚烯烃系树脂发泡片,沿厚度方向将上述聚烯烃系树脂发泡片5等分,在按厚度方向的顺序设为第1~5层时,关于第1层和第5层的凝胶分
率,如果将值大的一方设为gfa,将值小的一方设为gfb,则由gfa/gfb算出的表层的凝胶分率比为1.0以上且1.2以下。
26.(10)根据(1)~(9)中任一项所述的聚烯烃系树脂发泡片,沿厚度方向将上述聚烯烃系树脂发泡片5等分,在按厚度方向的顺序设为第1~5层时,关于第1层和第5层的平均气泡直径bd,如果将值大的一方设为bda,将值小的一方设为bdb,则由bda/bdb算出的表层的平均气泡直径比为1.0以上且1.2以下。
27.(11)根据(1)~(10)中任一项所述的聚烯烃系树脂发泡片,关于md方向和td方向两者,在将加热前的上述聚烯烃系树脂发泡片的平均气泡直径设为bd
bf
,将在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟的发泡片的平均气泡直径设为bd
af
时,由bd
bf
/bd
af
算出的加热前后的平均气泡直径比为1.0以上且1.5以下。
28.(12)一种叠层体,其是使选自片、膜、布、无纺织物和皮中的1种以上表皮材料、与(1)~(11)中任一项所述的聚烯烃系树脂发泡片叠层而得的。
29.发明的效果
30.根据本发明,可以提供兼有优异的柔软性和成型性的、聚烯烃系树脂发泡片及其叠层体。
附图说明
31.图1为说明本发明涉及的聚烯烃系树脂发泡片的平均气泡直径的测定的图。
具体实施方式
32.本发明涉及的聚烯烃系树脂发泡片将包含聚乙烯系树脂0质量%以上且30质量%以下、聚丙烯系树脂30质量%以上且80质量%以下和聚烯烃系弹性体20质量%以上且40质量%以下的树脂混合物作为基材树脂。
33.<基材树脂>
34.作为在本发明中使用的聚乙烯系树脂,为主要包含聚乙烯的树脂,可举出例如,高密度聚乙烯(hdpe)、低密度聚乙烯(ldpe)、直链状低密度聚乙烯(lldpe)、乙烯-丙烯酸乙酯共聚物(eea)、乙烯-丙烯酸丁酯共聚物(eba)等。此外,也可以根据需要使用乙烯单体与其它能够共聚的单体的共聚物。这些聚乙烯系树脂不仅可以为1种,也可以掺混2种以上。此外,对这些聚乙烯系树脂的聚合方法没有特别限制,可以为高压法、浆料法、溶液法、气相法中的任一者,关于聚合催化剂,也不特别限定于齐格勒催化剂、金属茂催化剂等。
35.聚乙烯系树脂没有特别限定,但优选使用密度在890kg/m3以上且950kg/m3以下,mfr(190℃)在1g/10分钟以上且15g/10分钟以下的范围内的物质,其中特别优选使用密度为920kg/m3以上且940kg/m3以下,mfr(190℃)为2g/10分钟以上且10g/10分钟以下,熔点为100℃以上且130℃以下的乙烯-α-烯烃共聚物。
36.聚乙烯系树脂在基材树脂中的比例为0质量%以上且30质量%以下。通过使聚乙烯系树脂为0质量%以上且30质量%以下,从而可以赋予优异的柔软性和成型性。如果聚乙烯系树脂超过30质量%,则成型时的收缩变大,发生尺寸缺损等不良状况。聚乙烯系树脂在基材树脂中的比例优选为0质量%以上且25质量%以下,更优选为0质量%以上且20质量%以下,进一步优选为0质量%以上且15质量%以下。
37.作为在本发明中使用的聚丙烯系树脂,是主要包含聚丙烯的树脂,可举出例如,均聚丙烯、乙烯-丙烯无规共聚物、乙烯-丙烯嵌段共聚物等。此外,也可以根据需要使用丙烯单体与其它能够共聚的单体的共聚物。聚烯烃系树脂发泡片中的聚丙烯系树脂不仅可以为1种,也可以掺混使用2种以上。此外,对这些聚丙烯系树脂的聚合方法没有特别限制,可以为高压法、浆料法、溶液法、气相法中的任一者,关于聚合催化剂,也不特别限定于齐格勒催化剂、金属茂催化剂等。
38.聚丙烯系树脂没有特别限定,但特别优选使用聚丙烯系树脂100质量%中的乙烯含有率为5质量%以上且15质量%以下,熔点为135℃以上且160℃以下,mfr(230℃)为0.5g/10分钟以上且5.0g/10分钟以下的无规聚丙烯、或聚丙烯系树脂100质量%中的乙烯含有率为1质量%以上且5质量%以下,熔点为150℃以上且170℃以下,mfr(230℃)为1.0g/10分钟以上且7.0g/10分钟以下的嵌段聚丙烯。
39.聚丙烯系树脂在基材树脂中的比例为30质量%以上且80质量%以下。通过使聚丙烯系树脂为30质量%以上且80质量%以下,从而可以赋予优异的柔软性和成型性。如果聚丙烯系树脂小于30质量%,则成型时的收缩变大,发生尺寸缺损等不良状况。如果聚丙烯系树脂超过80质量%,则不能赋予充分的柔软性。聚丙烯系树脂在基材树脂中的比例优选为30质量%以上且70质量%以下,更优选为30质量%以上且60质量%以下,进一步优选为30质量%以上且50质量%以下。
40.在本发明中使用的聚烯烃系弹性体多为由软链段和硬链段构成的物质,也可以根据需要使用乙烯单体和丙烯单体与其它能够共聚的单体的共聚物。这些聚烯烃系弹性体不仅可以为1种,也可以掺混2种以上。此外,对聚合方法没有特别限制,可以为高压法、浆料法、溶液法、气相法中的任一者,关于聚合催化剂,也不特别限定于齐格勒催化剂、金属茂催化剂等。也可以进一步物理混合2种以上的成为硬链段的聚合物与成为软链段的聚合物而制成聚合物合金。在不损害本发明的效果的范围,可以含有聚苯乙烯系弹性体(sbc、tps)、氯乙烯系弹性体(tpvc)、聚氨酯系弹性体(tpu)、聚酯系弹性体(tpee、tpc)、聚酰胺系弹性体(tpae、tpa)、聚丁二烯系弹性体等弹性体。
41.聚烯烃系弹性体没有特别限定,但优选使用熔点为120℃以上且160℃以下,mfr(230℃)为0.1g/10分钟以上且40.0g/10分钟以下,玻璃化转变温度为-40℃以下的聚烯烃系弹性体。
42.聚烯烃系弹性体的基材树脂中的比例为20质量%以上且40质量%以下。通过使聚烯烃系弹性体为20以上且40质量%以下,从而可以赋予优异的柔软性和成型性。如果聚烯烃系弹性体小于20质量%,则不能赋予充分的柔软性。如果聚烯烃系弹性体超过40质量%,则成型时的收缩变大,发生尺寸缺损等不良状况。聚烯烃系弹性体在基材树脂中的比例优选为20质量%以上且35质量%以下,更优选为25质量%以上且35质量%以下,进一步优选为30质量%以上且35质量%以下。
43.<发泡剂>
44.本发明的聚烯烃系树脂发泡片是在基材树脂中混合可以产生气体的发泡剂而制造的。作为其制造方法,可举出:在基材树脂中加入作为发泡剂的热分解型化学发泡剂进行熔融混炼,在常压加热下发泡的常压发泡法;在挤出机内将热分解型化学发泡剂加热分解,在高压下一边挤出一边发泡的挤出发泡法;在压制模具内将热分解型化学发泡剂加热分
解,一边减压一边发泡的压制发泡法;以及在挤出机内将气体或进行气化的溶剂熔融混合,在高压下一边挤出一边发泡的挤出发泡法等方法。
45.这里使用的所谓热分解型化学发泡剂,是通过施加热进行分解而放出气体的化学发泡剂,可举出例如,偶氮二甲酰胺、n,n
’‑
二亚硝基五亚甲基四胺、p,p
’‑
氧基苯磺酰肼等有机系发泡剂、碳酸氢钠、碳酸铵、碳酸氢铵和叠氮化钙等无机系发泡剂。
46.发泡剂可以分别单独使用或组合使用2种以上。为了获得柔软,成型性高,表面平滑的高倍率的发泡体,适合使用:使用了偶氮二甲酰胺作为发泡剂的常压发泡法。
47.<交联助剂>
48.本发明的聚烯烃系树脂发泡片可以使用被交联了的树脂发泡体(称为交联发泡体)、未被交联的树脂发泡体(称为非交联发泡体)中的任一者,只要根据用途来选择适当的树脂发泡体即可。从树脂发泡体的表面具有平滑性,叠层体的外观优异方面、在成型时不易破裂因此可以追求设计性方面考虑,聚烯烃系树脂发泡片优选为被交联了的树脂发泡体。用于制成交联发泡体的方法没有特别限制。作为获得交联发泡体的方法,可举出例如,通过使原料中含有具有硅烷基、过氧化物、羟基、酰胺基、酯基等化学结构的交联剂从而化学地交联的化学交联方法;通过将电子射线、α射线、β射线、γ射线、紫外线辐射到聚烯烃系树脂从而进行交联的放射线交联方法等。在仅通过电子射线照射难以构建交联结构的情况下,通过使用于制造聚烯烃系树脂发泡片的基材树脂中含有交联助剂从而可以通过电子射线而获得交联发泡体。作为交联助剂,没有特别限制,但优选使用多官能单体。作为多官能单体,可以使用例如,二乙烯基苯、三羟甲基丙烷三甲基丙烯酸酯、1,6-己二醇二甲基丙烯酸酯、1,9-壬二醇二甲基丙烯酸酯、1,10-癸二醇二甲基丙烯酸酯、偏苯三甲酸三烯丙酯、异氰脲酸三烯丙酯、乙基乙烯基苯等。这些多官能单体可以分别单独使用,或也可以组合使用2种以上。
49.<其它添加剂>
50.在基材树脂和聚烯烃系树脂发泡片中,根据需要,可以包含抗氧化剂、热稳定剂、着色剂、阻燃剂、抗静电剂等。
51.<混配比例>
52.在本发明涉及的聚烯烃系树脂发泡片的基材树脂100质量%中,以聚乙烯系树脂0质量%以上且30质量%以下,聚丙烯系树脂30质量%以上且80质量%以下,聚烯烃系弹性体20质量%以上且40质量%以下的比例混配。
53.<聚烯烃系树脂发泡片>
54.本发明涉及的聚烯烃系树脂发泡片优选为独立气泡结构。在独立气泡结构的发泡体的情况下,由于其结构因此在真空成型中可以将空气充分地抽出等,能够进行向复杂形状的成型。此外,从发泡体、成型了发泡体的成型品的表面变得平滑考虑,优选气泡微细而均匀。
55.在使用本发明涉及的聚烯烃系树脂发泡片作为汽车内装材料的情况下,聚烯烃系树脂发泡片的厚度优选为1.0mm以上且5.0mm以下。如果厚度小于1.0mm,则可能发生触底。此外,如果厚度超过5.0mm则作为构件的轻量性变差。厚度更优选为1.0mm以上且4.0mm以下,进一步优选为2.0mm以上且4.0mm以下。
56.本发明涉及的聚烯烃系树脂发泡片的表观密度优选为40kg/m3以上且100kg/m3以
下。如果表观密度小于40kg/m3则可能发生触底,如果超过100kg/m3则不能赋予充分的柔软性。聚烯烃系树脂发泡片的表观密度更优选为50kg/m3以上且100kg/m3以下,进一步优选为50kg/m3以上且80kg/m3以下。
57.本发明中所谓凝胶分率,是基材树脂之中的被交联而被高分子化了的树脂的比例,是指在通常被成型的温度下不增塑的部分的比例。一般而言如果该部分变多则耐热性提高,但成型性降低。因此,根据成型工艺而任意地选择该比率。本发明涉及的聚烯烃系树脂发泡片的凝胶分率优选为30%以上且60%以下。如果凝胶分率小于30%,则耐热性降低而在成型加工时发泡片劣化,难以成型加工。此外,如果凝胶分率超过60%,则可能损害柔软性。聚烯烃系树脂发泡片的凝胶分率更优选为30%以上且55%以下,进一步优选为30%以上且50%以下。
58.此外,将本发明的聚烯烃系树脂发泡片沿厚度方向5等分,在按厚度方向的顺序设为第1~5层时,关于第1层和第5层的凝胶分率,如果将值大的一方设为gfa,将值小的一方设为gfb,则由gfa/gfb算出的表层的凝胶分率比优选为1.0以上且1.2以下。通过使上述表层凝胶分率比为1.0以上且1.2以下,从而可以赋予优异的成型性。如果表层的凝胶分率比超过1.2,则发泡体的卷曲变大,发生尺寸缺损、褶皱引起的外观不良等成型不良状况。表层的凝胶分率比更优选为1.0以上且1.1以下。
59.本发明涉及的聚烯烃系树脂发泡片的25%压缩强度优选为250kpa以下。如果25%压缩强度超过250kpa则难以赋予充分的柔软性。25%压缩强度更优选为200kpa以下,进一步优选为150kpa以下。
60.在本发明涉及的聚烯烃系树脂发泡片中,将25%压缩强度(kpa)除以密度(kg/m3)而得的值优选为2.5以下。如果将25%压缩强度(kpa)除以密度(kg/m3)而得的值超过2.5则难以赋予充分的柔软性。将25%压缩强度(kpa)除以密度(kg/m3)而得的值更优选为2.3以下,进一步优选为2.1以下,特别优选为1.9以下。
61.本发明涉及的聚烯烃系树脂发泡片的23℃下的抗拉强度(md方向、td方向)优选为500kpa以上。如果23℃下的抗拉强度(md方向、td方向)小于500kpa,则可能在成型加工时发生破裂而不能获得良好的成型品。23℃下的抗拉强度(md方向、td方向)更优选为700kpa以上,进一步优选为900kpa以上。
62.在本发明涉及的聚烯烃系树脂发泡片中,将23℃下的md方向抗拉强度除以td方向抗拉强度而得的、抗拉强度比优选为0.7以上且1.3以下。如果抗拉强度比小于0.7或超过1.3,则由成型加工时的加热引起的收缩变大,可能发生尺寸缺损而不能获得成型体。抗拉强度比更优选为0.8以上且1.3以下,进一步优选为0.8以上且1.2以下,特别优选为0.9以上且1.1。
63.本发明涉及的聚烯烃系树脂发泡片的-35℃下的抗拉强度(md方向、td方向)优选为500kpa以上。如果-35℃的抗拉强度(md方向、td方向)小于500kpa,则可能在成型加工时发生破裂而不能获得良好的成型品。-35℃下的抗拉强度(md方向、td方向)更优选为700kpa以上,进一步优选为900kpa以上。
64.本发明涉及的聚烯烃系树脂发泡片的23℃下的拉伸伸长率(md方向、td方向)优选为200%以上。如果23℃下的拉伸伸长率(md方向、td方向)小于200%,则可能在成型加工时发生破裂而不能获得良好的成型品。23℃的拉伸伸长率(md方向、td方向)更优选为250%以
上,进一步优选为300%以上。
65.本发明涉及的聚烯烃系树脂发泡片的-35℃下的拉伸伸长率(md方向、td方向)优选为30%以上。如果-35℃下的拉伸伸长率(md方向、td方向)小于30%,则可能在成型加工时发生破裂而不能获得良好的成型品。-35℃下的拉伸伸长率(md方向、td方向)更优选为40%以上,进一步优选为50%以上。
66.本发明涉及的聚烯烃系树脂发泡片的23℃下的撕裂强度(md方向、td方向)优选为50n/cm以上。如果23℃下的撕裂强度(md方向、td方向)小于50n/cm,则可能在成型加工时发生破裂而不能获得良好的成型品。23℃下的撕裂强度(md方向、td方向)优选为60n/cm以上,更优选为70n/cm以上。
67.在本发明涉及的聚烯烃系树脂发泡片中,将23℃下的md方向撕裂强度除以td方向撕裂强度而得的、撕裂强度比优选为0.7以上且1.3以下。如果抗拉强度比小于0.7或超过1.3,则由成型加工时的加热引起的收缩变大,可能发生尺寸缺损而不能获得成型体。撕裂强度比更优选为0.8以上且1.3以下,进一步优选为0.8以上且1.2以下,特别优选为0.9以上且1.1以下。
68.在本发明涉及的聚烯烃系树脂发泡片中,在120℃下进行了1小时加热时的加热尺寸变化率(md方向、td方向)优选为-5%以上0%以下。通过加热尺寸变化率在该范围,从而可以抑制加热成型时的收缩,获得良好的成型体。md方向和td方向的加热尺寸变化率更优选为-4%以上且0%以下,进一步优选为-3%以上且0%以下。
69.在本发明涉及的聚烯烃系树脂发泡片中,在比在dsc测定中作为最高熔融峰的最高熔点低20℃的温度下加热了10分钟时的加热尺寸变化率(md方向、td方向)优选为-5%以上且0%以下。通过使上述加热尺寸变化率为-5%以上且0%以下,从而加热成型时的收缩被抑制,可以防止尺寸缺损等成型不良。比md方向和td方向的最高熔点低20℃的温度下的加热尺寸变化更优选为-4%以上且0%以下,进一步优选为-3以上且0%以下。
70.在本发明涉及的聚烯烃系树脂发泡片中,在比在dsc测定中作为最高熔融峰的最高熔点低20℃的温度下加热了10分钟时的将md方向的加热尺寸变化率dc
md
除以td方向的加热尺寸变化率dc
td
而得的、加热尺寸变化率比dc
md
/dc
td
优选为0.5以上且1.5以下。通过比最高熔点低20℃的温度下的加热尺寸变化率比dc
md
/dc
td
在该范围内,从而可以减轻加热成型时的收缩各向异性,获得良好的成型品。比最高熔点低20℃的温度的加热尺寸变化率比dc
md
/dc
td
更优选为0.7以上且1.5以下,进一步优选为0.7以上且1.4以下,特别优选为0.8以上且1.3以下。
71.在本发明涉及的聚烯烃系树脂发泡片中,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的加热尺寸变化率(md方向、td方向)为-35%以上且0%以下。通过使上述加热尺寸变化率为-35%以上且0%以下,从而加热成型时的收缩被抑制,可以防止尺寸缺损等成型不良。比最高熔点高20℃的温度下的加热尺寸变化优选为-33%以上,更优选为-31%以上,进一步优选为-30%以上。
72.在本发明涉及的聚烯烃系树脂发泡片中,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的将md方向的加热尺寸变化率dc
md
除以td方向的加热尺寸变化率dc
td
而得的、加热尺寸变化率比dc
md
/dc
td
优选为0.5以上且1.5以下。通过比最高熔点高20℃的温度下的加热尺寸变化率比dc
md
/dc
td
在该范围内,从而可以减轻加热成型
时的收缩各向异性,获得良好的成型品。比最高熔点高20℃的温度下的加热尺寸变化率比dc
md
/dc
td
更优选为0.6以上且1.4以下,进一步优选为0.7以上且1.3以下。
73.在本发明涉及的聚烯烃系树脂发泡片中,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的卷曲高度优选为发泡片厚度以上且15mm以下。通过使上述卷曲高度为发泡片厚度以上15mm以下,从而可以赋予优异的成型性。如果卷曲高度超过15mm,则由尺寸缺损、褶皱引起的外观不良等成型不良状况发生。优选卷曲高度低,但发泡片的厚度成为实质的下限。聚烯烃系树脂发泡片的卷曲高度更优选为发泡片厚度以上且14mm以下,进一步优选为发泡片厚度以上且13mm以下,特别优选为发泡片厚度以上且12mm以下。
74.可以通过使聚烯烃系树脂发泡片的表层的凝胶分率比小而使聚烯烃系树脂发泡片的卷曲高度低。表层的凝胶分率比是,将聚烯烃系树脂发泡片沿厚度方向5等分,在按厚度方向的顺序设为第1~5层时,作为表层的第1层和第5层的凝胶分率之中,如果将值大的一方设为gfa,将值小的一方设为gfb,由gfa/gfb算出的值。
75.此外,通过使聚烯烃系树脂发泡片的表层的平均气泡直径比小,从而可以使聚烯烃系树脂发泡片的卷曲高度低。表层的平均气泡直径比是,第1层与第5层的平均气泡直径之中,如果将值大的一方设为bda,将值小的一方设为bdb,由bda/bdb算出的值。
76.进一步,使基材树脂中的聚乙烯系树脂、聚烯烃系树脂的比例在不损害柔软性的范围小也具有使卷曲高度低的效果。可以通过调整树脂组成、表层的凝胶分率比、表层的平均气泡直径比中的任一者、或多个而使卷曲高度低,优选对多个进行调整。
77.本发明涉及的聚烯烃系树脂发泡片的平均气泡直径(md方向、td方向)优选为50μm以上且500μm以下。如果平均气泡直径小于50μm,则可能耐热性降低。如果平均气泡直径超过500μm,则失去表面的平滑性,在成型时可能发生凹陷。聚烯烃系树脂发泡片的平均气泡直径更优选为100μm以上且500μm以下,进一步优选为200μm以上且500μm以下。
78.本发明涉及的聚烯烃系树脂发泡片的将md方向的平均气泡直径bd
md
除以td方向的平均气泡直径bd
td
而得的平均气泡直径比bd
md
/bd
td
优选为0.7以上且1.3以下。如果平均气泡直径比小于0.7或超过1.3,则由成型加工时的加热引起的收缩变大,可能发生尺寸缺损而不能获得成型体。聚烯烃系树脂发泡片的平均气泡直径比bd
md
/bd
td
更优选为0.8以上且1.3以下,进一步优选为0.8以上且1.2以下,特别优选为0.9以上且1.1以下。在生产工序中,如果拉伸应力沿md方向作用,则残余应力残留因此扁平的气泡沿md方向产生。此外,在发泡工序中被加热时,通过发泡剂的分解而形成的气泡要变为圆形,但如果施加应力则气泡变为扁平状态。通过气泡的扁平程度,可以判别生产时的拉伸应力的强弱,因此平均气泡直径比bd
md
/bd
td
小的发泡体的加热尺寸收缩变小,具有优异的成型性。
79.本发明涉及的聚烯烃系树脂发泡片的表层的平均气泡直径比优选为1.0以上且1.2以下。表层的平均气泡直径比是,沿厚度方向将聚烯烃系树脂发泡片5等分,在按厚度方向的顺序设为第1~5层时,关于第1层和第5层的平均气泡直径bd,如果将值大的一方设为bda,将值小的一方设为bdb,由bda/bdb算出的值。通过使表层的平均气泡直径比bda/bdb为1.0以上且1.2以下,从而可以减轻发泡体的卷曲,防止由尺寸缺损、褶皱引起的外观不良等成型不良。表层的平均气泡直径比bda/bdb更优选为1.0以上且1.1以下,进一步优选为1.0。
80.在本发明涉及的聚烯烃系树脂发泡片中,加热前的平均气泡直径bd
bf
和在比在dsc
测定中作为最高熔融峰的最高熔点高20度的温度下加热10分钟后的平均气泡直径bd
af
之比bd
bf
/bd
af
(md方向、td方向)优选为1.0以上且1.5以下。通过使加热前后的平均气泡直径比bd
bf
/bd
af
为1.0以上且1.5以下,从而可以赋予优异的成型性。如果加热前后的平均气泡直径比bd
bf
/bd
af
超过1.5,则可能发生尺寸缺损等成型不良状况。聚烯烃系树脂发泡片的加热前后的平均气泡直径比bd
bf
/bd
af
更优选为1.0以上且1.4以下,进一步优选为1.0以上且1.3以下,特别优选为1.0以上且1.2以下。
81.<叠层体>
82.本发明涉及的叠层体是使选自片、膜、布、皮等中的1种以上表皮材料、与上述聚烯烃系树脂发泡片叠层而得的。通过在本发明的聚烯烃系树脂发泡片叠层表皮材料,从而能够通过良好设计性而赋予高级感等。对表皮材料的材质没有特别限制,可举出例如,包含聚乙烯、聚丙烯、乙烯-乙酸乙烯酯共聚物(eva)、乙烯-丙烯酸乙酯共聚物(eea)、乙烯-丙烯酸丁酯共聚物(eba)、乙烯-丙烯橡胶等弹性体成分的热塑性聚烯烃系弹性体(tpo)的片、膜、聚氯乙烯、聚偏1,1-二氯乙烯等乙烯基树脂、聚氨酯树脂、聚苯乙烯系树脂、聚醚系树脂、聚酰胺树脂、由这些树脂与能够共聚的单体构成的共聚物的片、膜、布、无纺织物、或皮等。这些表皮材料可以使至少1种或2种以上混合使用。
83.<聚烯烃系树脂发泡片的制造方法>
84.本发明的聚烯烃系树脂发泡片可以通过将基材树脂成型为片状而获得发泡性片的工序、将上述发泡性片交联的工序、将交联了的发泡性片加热发泡而获得发泡片的工序来制造。以下,以使用了热分解型发泡剂作为发泡剂的常压发泡法作为例子,对本发明的聚烯烃系树脂发泡片的制造方法进行说明。
85.获得发泡性片的工序是,将由聚乙烯系树脂、聚丙烯系树脂、烯烃系弹性体等构成的基材树脂、和热分解型发泡剂使用亨舍尔混合机、转筒等混合设备均匀地混合。然后,使用挤出机、加压式捏合机等熔融混炼设备,在小于热分解型发泡剂的分解温度的温度条件下均匀地熔融混炼,通过t型口模而成型为片状。在成型为片状时,优选使牵伸率降低,即在减少了拉伸应力的状态下成型。作为牵伸率,是由片厚度相对于口模前端的间隙之比计算的数值,数值越小则表示从口模被挤出的发泡性片越不被拉伸。通过使牵伸率小,从而可以减少发泡性片的md方向的应变,也可以减少残存于发泡片的应变,因此可以使成型加热时的收缩小,即,防止尺寸缺损而提高成型性。通常,与获得发泡性片的工序的成型温度相比,获得发泡片的工序的发泡温度高,因此如果牵伸率大,发泡性片的应变大地残留,则在发泡初始发生应变的松弛而沿md方向收缩。md方向拉伸倍率是将卷绕速度除以开卷速度而算出,但通过上述收缩而实际的开卷速度成为慢的状态,成为沿md方向更被拉伸了的状态。此外,如果由应变的松弛引起的md方向的收缩大,则发泡状态不稳定,因此难以减少设定上的md方向拉伸倍率。此外,对于表示口模与将从口模被排出的片成型的最初的轧辊之间的距离的所谓气隙而言,根据排出的树脂的量、片的厚度、宽度而有所不同,但优选扩大。通过扩大气隙,从而能够将口模以后的树脂的取向松弛。因此,通过在垂伸、向内弯曲被容许的范围,具有充分的距离,从而能够减少发泡性片的应变,可以减少发泡片的收缩。此外,成型片时的温度也在热分解型发泡剂不分解的范围,设定得高时可以减少应变因此是优选的,从口模被排出的基材树脂的温度优选为165℃以上且190℃以下的范围。进一步,使将成型出的片卷绕时的张力也降低至片不卷崩溃的程度是重要的。在将基材树脂与热分解型发泡剂
混合时,根据需要,可以添加抗氧化剂、热稳定剂、交联助剂等。
86.将发泡性片交联的工序是,向成型出的发泡性片照射电离放射线,将发泡性片交联。作为电离放射线,可举出例如,电子射线、α射线、β射线、γ射线、x射线等,如果考虑生产性则优选使用电子射线。
87.获得发泡片的工序是,使交联了的发泡性片加热发泡,获得聚烯烃系树脂发泡片。具体而言,通过加热而使基材树脂软化,并且使其升温到热分解型发泡剂的分解温度以上,通过由热分解型发泡剂的分解而产生的气体而使基材树脂发泡,从而可以获得本发明的聚烯烃系树脂发泡片。作为加热方法,可举出在成为热媒的盐浴上浮起的方法、投入到热风等环境下中投入的方法。通过极力减少在发泡中施加的应力而抑制应变,从而可以提高将聚烯烃系树脂发泡片加热成型时的加热尺寸收缩,即成型性,因此优选为在盐浴上浮起的方法。此外,交联了的发泡性片可以沿md方向和/或td方向被拉伸。作为考虑了生产性的实施方法,可举出将卷状的交联了的发泡性片连续地供给到高温的盐浴,并卷绕为卷状的制品。此时,将卷绕速度除以开卷速度而得的md方向拉伸倍率优选为2.0以上且3.0以下。如果md方向拉伸倍率小于2.0,则可能发泡过程的片曲折而得不到良好的发泡片。另一方面,如果md方向拉伸倍率超过3.0,则施加于发泡片的应力变大因此可能在发泡片残留应变,成型加工时的加热尺寸收缩变大,即发生尺寸缺损而不能成型。md方向拉伸倍率优选为2.2以上且2.8以下,更优选为2.2以上且2.7以下,进一步优选为2.3以上且2.7以下。为了减轻交联了的发泡性片的应变而使发泡状态稳定,在加热到发泡剂的分解温度以上之前,优选进行预加热。作为预加热时的温度,优选为在包含聚乙烯系树脂、聚丙烯系树脂、和聚烯烃系弹性体的树脂混合物的在dsc测定中获得的最高熔融峰温度以下,并且比最低熔融峰温度低30℃的温度以上。通过在该温度范围将发泡性片预加热从而能够减少片的应变,能够减少发泡工序中的md方向拉伸倍率。进一步,关于发泡时的加热温度,由于能够通过使发泡缓慢从而降低md方向拉伸倍率,因此优选不是恒定的温度,而是在发泡的前半部与后半部的温度设置温度差。此外,从减少发泡体的md方向的收缩的观点考虑,在发泡工序中,对于在将发泡体冷却后到卷绕之前的输送辊,优选减少辊的旋转阻力等,使md方向拉伸倍率降低。将树脂发泡片的td方向长度除以发泡前的树脂发泡性片的td方向长度而得的td方向拉伸倍率优选与md方向拉伸倍率同等。
88.<叠层体的制造方法>
89.在聚烯烃系树脂发泡片叠层表皮材料而制成叠层体的方法没有特别限制,可举出挤出层压法、粘接层压法、热层压法、热熔法等。
90.<聚烯烃系树脂发泡片或叠层体的成型>
91.本发明的聚烯烃系树脂发泡片或叠层体的成型方法没有特别限制,可举出挤出成型、真空成型、冲压成型、吹塑成型等公知的方法。通过这些方法而获得的成型品可以通过热熔接、振动熔接、超声波熔接、激光熔接等而二次加工为根据需要的形状。
92.实施例
93.<物性评价>
94.按照下述方法测定了在发泡后在至少4天以上、温度23℃、湿度50%的条件下养护了的聚烯烃系树脂发泡片的各种物性。另外,md方向表示长度方向,td方向表示宽度方向。在不能判别md方向和td方向的情况下,将气泡的直径最长的方向作为md方向,将其垂直方
向作为td方向而对待。
95.关于本发明的物性范围,在没有限定于md方向或td方向的记载的情况下,需要md方向和td方向两者满足范围条件。此外,关于物性值,将所得的值四舍五入,由说明书记载的有效数字判断。
96.(1)厚度(mm)
97.聚烯烃系树脂发泡片的厚度按照iso1923:1981“发泡塑料和橡胶-线尺寸的测定方法(発泡
プラスチック
及
びゴムー
線寸法
の
測定方法)”而测定。具体而言,使树脂发泡片静置在平坦的台,使带有具有10cm2的面积的圆形测头的直读式厚度计与树脂发泡片表面以10g/10cm2的恒定压力接触而测定。
98.(2)表观密度(kg/m3)
99.聚烯烃系树脂发泡片的表观密度按照jisk6767:1999“发泡塑料-聚乙烯-试验方法(発泡
プラスチックーポリエチレンー
試験方法)”而测定。具体而言,测定10cm见方的试验片(聚烯烃系树脂发泡片)的厚度和质量,由下式算出。
100.密度(kg/m3)=试验片的质量(kg)/[试验片面积0.0001(m2)
×
试验片的厚度(m)]
[0101]
(3)发泡倍数(cm3/g)
[0102]
关于聚烯烃系树脂发泡片的发泡倍数,将按照jisk6767:1999“发泡塑料-聚乙烯-试验方法(発泡
プラスチック
-
ポリエチレン
-試験方法)”而测定的、表观密度的倒数设为发泡倍数。
[0103]
(4)凝胶分率、表层的凝胶分率比(%)
[0104]
将聚烯烃系树脂发泡片切断为约0.5mm见方,将切断了的聚烯烃系树脂发泡片以0.1mg单位称量约100mg。在130℃的温度的四氢化萘200ml中浸渍称量了的聚烯烃系树脂发泡片3小时后,用100目的不锈钢制金属网进行自然过滤,将金属网上的不溶解成分在温度120℃、1小时的条件下利用热风炉进行干燥。接着,在加入了被干燥了的硅胶的干燥器内冷却10分钟,将该不溶解成分的质量以0.1mg单位称量,按照下式,将凝胶分率以百分率而算出。
[0105]
凝胶分率(%)=[不溶解成分的质量(mg)/称量了的发泡体的质量(mg)]
×
100
[0106]
表层的凝胶分率如以下那样算出。将聚烯烃系树脂发泡片,使用切片机(株式会社
ニッピ
機械制np-120rs)沿厚度方向5等分,按厚度方向的顺序设为第1~5层。关于第1层和第5层的发泡体,与上述凝胶分率的测定同样地操作而求出凝胶分率,在将值大的一方设为gfa,将值小的一方设为gfb时,将由gfa/gfb算出的值设为表层的凝胶分率比。
[0107]
(5)25%压缩应力(kpa)
[0108]
聚烯烃系树脂发泡片的25%压缩应力按照jisk6767:1999“发泡塑料-聚乙烯-试验方法(発泡
プラスチック
-
ポリエチレン
-試験方法)”而测定。具体而言,将聚烯烃系树脂发泡片切断为50mm
×
50mm,将切断了的聚烯烃系树脂发泡片以厚度成为20mm以上且30mm以下的方式叠层,测定初始厚度。在平面板放置叠层了的样品,以10mm/分钟的速度压缩直到初始厚度的25%,停止,测定20秒后的荷重,通过下式而算出。
[0109]
25%压缩应力(kpa)=25%压缩后20秒后的荷重(n)/0.0025(m2)/1000
[0110]
(6)抗拉强度(kpa)/拉伸伸长率(%)
[0111]
聚烯烃系树脂发泡片的抗拉强度和拉伸伸长率按照jisk6767:1999“发泡塑料-聚
乙烯-试验方法(発泡
プラスチック
-
ポリエチレン
-試験方法)”而测定。将聚烯烃系树脂发泡片以md方向和td方向分别成为长度方向的方式用哑铃模冲裁而制作出试验片。
[0112]
将试验片在被调整为23℃的恒温槽内静置5分钟后,在23℃环境下实施了单轴拉伸试验。将此时的、强度的最大值设为23℃抗拉强度,将达到断裂时的伸长率设为23℃拉伸伸长率。将md方向的抗拉强度ts
md
除以td方向的抗拉强度ts
td
而得的值设为抗拉强度比ts
md
/ts
td
,将md方向的拉伸伸长率te
md
除以td方向的拉伸伸长率te
td
而得的值设为拉伸伸长率比te
md
/te
td
。
[0113]
此外,将试验片在被调整为-35℃的恒温槽内静置5分钟后,在-35℃环境下实施了单轴拉伸试验。将此时的、强度的最大值设为-35℃抗拉强度,将达到断裂时的伸长率设为-35℃拉伸伸长率。
[0114]
(7)撕裂强度(n/cm)
[0115]
聚烯烃系树脂发泡片的撕裂强度按照jisk6767:1999“发泡塑料-聚乙烯-试验方法(発泡
プラスチック
-
ポリエチレン
-試験方法)”而测定。将聚烯烃系树脂发泡片以md方向和td方向分别成为长度方向的方式用模冲裁而制作出试验片。这里,md方向表示流动方向,td方向表示宽度方向。将试验片在被调整为23℃的恒温槽内静置5分钟后,在23℃环境下实施了撕裂试验。将此时的、切断时的最大荷重设为撕裂强度。将md方向的撕裂强度tes
md
除以td方向的撕裂强度tes
td
而得的值设为撕裂强度比tes
md
/tes
td
。
[0116]
(8)加热尺寸变化率(%)
[0117]
聚烯烃系树脂发泡片的加热尺寸变化按照jisk7133:1999“塑料-膜和片-加热尺寸变化测定方法(
プラスチック
-
フィルム
及
びシート
-加熱寸法変化測定方法)”而测定。具体而言,将聚烯烃系树脂发泡片的td方向中心以md方向与2边成为平行的方式冲裁为120
×
120mm的正方形,制作出试验片。沿试验片的md方向和td方向画出标线,使用游标卡尺以0.1mm单位测长。接下来,将加入了高岭土床的金属制容器放入120℃的烘箱中,将高岭土床调整为120℃。在试验片撒上高岭土,平坦地放置在高岭土床上,在120℃下加热了1小时。在加热后,在温度23℃、湿度50%的环境下冷却30分钟以上,将试验后的md方向和td方向的标线长度,使用游标卡尺以0.1mm单位测长。由下式算出md方向和td方向的加热尺寸收缩。
[0118]
md加热尺寸变化率(dc
md
)=[(加热后md标线长度)-(加热前md标线长度)]/(加热前md标线长度)
×
100
[0119]
td加热尺寸变化率(dc
td
)=[(加热后td标线长度)-(加热前td标线长度)]/(加热前td标线长度)
×
100
[0120]
关于“比最高熔点高20℃的温度”和“比最高熔点低20℃的温度”,和除将加热时间从1小时变更为10分钟以外,也与加热温度同样地操作而测定。将md方向的加热尺寸变化率dc
md
除以td方向的加热尺寸变化率dc
td
而得的值设为加热尺寸变化率比dc
md
/dc
td
。
[0121]
(9)平均气泡直径(μm)、平均气泡直径比、表层的平均气泡直径比、加热前后的平均气泡直径比
[0122]
聚烯烃系树脂发泡片的平均气泡直径关于md方向和td方向分别测长而算出。关于平均气泡直径的测长,首先,将聚烯烃系树脂发泡片用剃刀裁切,制作与md方向平行的气泡截面开口的面,利用扫描型电子显微镜(株式会社日立
ハイテクノロジーズ
制s-3000n)将截面以任意的图像倍率拍摄。将所得的图像印刷在a4用纸上。图1为说明聚烯烃系树脂发泡
片的平均气泡直径的测定的图。如图1所示那样,在厚度方向的中心画出沿md方向气泡20个以上相接的任意直线,从直线的长度和与该直线相接的气泡数,通过下式而算出平均弦长。另外,上述任意直线尽可能不通过相邻的气泡间的接触点,而是通过气泡内部。此外,在上述直线通过气泡间的接触点的情况下,在通过该接触点的位置,将直线上的气泡数作为2而数出。
[0123]
平均弦长(μm)=直线的长度(μm)/气泡数(个)
[0124]
由算出的平均弦长,由下式算出md方向的平均气泡直径bd
md
。
[0125]
平均气泡直径(μm)=平均弦长(μm)/0.62
[0126]
在td方向中,也与md方向同样地操作而算出平均气泡直径bd
td
。
[0127]
将md方向的平均气泡直径bd
md
除以td方向的平均气泡直径bd
td
而得的值设为平均气泡直径比bd
md
/bd
td
。
[0128]
聚烯烃系树脂发泡片的表层的平均气泡直径比如以下那样算出。将聚烯烃系树脂发泡片使用切片机沿厚度方向5等分,按厚度方向的顺序设为第1~5层。关于第1层的发泡体,与上述平均气泡直径的测定同样地操作,在厚度方向中心画出直线,算出md方向和td方向的平均气泡直径,将它们的平均值设为第1层的平均气泡直径。关于第5层的发泡体,与上述平均气泡直径的测定同样地操作而算出md方向和td方向的平均气泡直径,将它们的平均值设为第5层的平均气泡直径。关于第1层和第5层的平均气泡直径,在将值大的一方设为bda,将值小的一方设为bdb时,将由bda/bdb算出的值设为表层的平均气泡直径比。
[0129]
加热前后的平均气泡直径比如以下那样算出。将加入了高岭土床的金属制容器加入比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度的烘箱进行调整。在发泡后在至少4天以上、温度23℃、湿度50%的条件下养护了的聚烯烃系树脂发泡片撒上高岭土,平坦地放置在高岭土床上,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟。加热后,在温度23℃、湿度50%的环境下冷却30分钟以上后,对于所得的聚烯烃系树脂发泡片,分别针对md方向和td方向,与上述平均气泡直径的测定同样地操作,在厚度方向中心画出直线,求出平均气泡直径,将其设为加热后的平均气泡直径bd
af
。关于md方向和td方向各自,将加热前的平均气泡直径设为bd
bf
,在将加热后的平均气泡直径设为bd
af
时,将由bd
bf
/bd
af
算出的值设为加热前后的平均气泡直径比bd
bf
/bd
af
。
[0130]
(10)最高熔点(℃)
[0131]
使用差示扫描量热计(dsc,
セイコー
電子工業株式会社制rdc220
‑ロボット
dsc)而测定。将聚烯烃系树脂发泡片5mg在氮气气氛下从室温以10℃/分钟的速度升温直到200℃后,在200℃下保持了5分钟(第1次运行)。接着,以10℃/分钟的速度冷却直到0℃后,再次以10℃/分钟的速度升温直到200℃(第2次运行)。读取第2次运行的最高温侧的熔融峰(吸热峰)的顶点的值,将其设为最高熔点。
[0132]
(11)卷曲高度(mm)
[0133]
使用比最高熔点高20℃的温度下的加热尺寸变化率测定后的试验片而测长。以发泡体试验片与金属板的接触面积变为最大的方式,将试验片放置在金属板上。在金属板面的垂直方向上,将发泡片的高度用游标卡尺测长,将最高点设为卷曲高度。
[0134]
(12)成型评价
[0135]
将聚烯烃系树脂发泡片以成为与md方向或td方向平行的方式切割,制作出200mm
见方的试验片。关于与md方向平行的2边,分别将距端部10mm的区域均等地夹住,进行了固定。以发泡片的两面的表面温度通过50秒~70秒的加热而成为比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度的方式,用红外线加热器加热,用具有150mm见方且深度20mm的真空孔的金属模进行了真空成型。另外,金属模以成为发泡片面的中心的方式放置,以发泡片与金属模的边成为平行的方式调整了位置。对于将与td方向平行的2边夹住了的试验片,也同样地进行了成型。成型评价利用目视以下述基准进行了5阶段评价。成型评价的值越大则表示成型性越优异,将成型评价3~5设为合格。另外,需要md方向和td方向两者满足下述评价基准。
[0136]
成型评价1:具有尺寸缺损,因为发泡片端部的折曲、褶皱而外观大幅差
[0137]
成型评价2:具有尺寸缺损,因为发泡片端部的折曲、褶皱而外观差
[0138]
成型评价3:没有尺寸缺损,可以确认发泡片端部的折曲、轻微的褶皱
[0139]
成型评价4:没有尺寸缺损,可以确认轻微的褶皱
[0140]
成型评价5:没有尺寸缺损,外观良好
[0141]
<使用树脂和添加剂>
[0142]
在实施例和比较例中,使用下述树脂和添加剂而实施。
[0143]
聚乙烯系树脂:日本
ポリエチレン
制,商品名
“ノバテック
(注册商标)uj960(mfr:5g/10分钟,密度:935kg/m3)”[0144]
聚丙烯系树脂:
サンアロマー
制,商品名“pb222a(mfr0.75g/10分钟,密度:900kg/m3)”[0145]
聚烯烃系弹性体:dow制,商品名“infuse(注册商标)9107(mfr:1g/10分钟,密度:866kg/m3)”[0146]
发泡剂:偶氮二甲酰胺(永和化成工业制,商品名
“ビニホール
(注册商标)ac#r”)
[0147]
交联助剂:55%二乙烯基苯(和光纯药工业制)
[0148]
抗氧化剂:basf制,商品名“irganox(注册商标)1010”[0149]
<实施例1~10、比较例1、4~6>
[0150]
对将聚乙烯系树脂、聚丙烯系树脂和聚烯烃系弹性体以表1记载的比例混合了的基材树脂100质量份,将发泡剂、交联助剂和抗氧化剂按照表1记载的添加量而添加了的混合物投入到亨舍尔混合机,进行了粉碎混合。
[0151]
将所得的混合物投入到双轴挤出机,在树脂温度160℃以上且180℃以下进行了熔融混炼后,使用t型模,以牵伸率1.4,成型为厚度1.4mm的片状,获得了卷绕成卷状的发泡性片。然而,为了调整发泡体的厚度,实施例3中使发泡性片的厚度为2.0mm,实施例4中使发泡性片的厚度为1.3mm,实施例5中使发泡性片的厚度为1.6mm。
[0152]
向所得的发泡性片,在加速电压800kv的条件下将照射剂量90kgy的电子射线从一面照射,获得了交联了的发泡性片。然而,为了调整发泡体的凝胶分率,实施例6中使照射剂量为60kgy,实施例7中使照射剂量为140kgy。
[0153]
将卷状的交联了的发泡性片用温水预加热到80℃以上且95℃以下后,连续地在调整为前半220℃以上且229℃以下、后半230℃以上且235℃以下的盐浴上浮起而加热,并且从上方也用红外线加热器加热,从而获得了聚烯烃系树脂发泡片。将发泡完成而从盐浴取出的卷绕速度除以供给到盐浴的开卷速度而得的md方向拉伸倍率调整为2.7。然而,为了调
整发泡体厚度,实施例4中使md方向拉伸倍率为3.0,实施例5中使md方向拉伸倍率为2.3。所得的发泡片在用50℃的水进行了冷却和洗涤后,用暖风干燥了。
[0154]
将所得的聚烯烃系树脂发泡片的物性示于表1~表3中。
[0155]
<实施例11>
[0156]
使牵伸率为1.6,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0157]
<实施例12>
[0158]
对将聚乙烯系树脂、聚丙烯系树脂和聚烯烃系弹性体以表1记载的比例混合了的基材树脂100质量份,将发泡剂、交联助剂和抗氧化剂按照表1记载的添加量而添加了的混合物投入到亨舍尔混合机,进行了粉碎混合。
[0159]
将所得的混合物投入到双轴挤出机,在树脂温度160℃以上且180℃以下进行了熔融混炼后,使用t型模,以牵伸率1.4,成型为厚度1.4mm的片状,获得了卷状地卷绕了的发泡性片。
[0160]
向所得的发泡性片,在加速电压800kv的条件下将照射剂量90kgy的电子射线从一面照射,获得了交联了的发泡性片。
[0161]
将卷状的交联了的发泡性片切断为10cm见方的尺寸,在调整为230以上且240℃以下的盐浴上浮起而加热,并且从上方注入上述温度的盐热媒,进行两面加热从而获得了聚烯烃系树脂发泡片。所得的发泡片用50℃的水进行了冷却和洗涤后,用暖风干燥了。
[0162]
将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0163]
<实施例13>
[0164]
使牵伸率为1.0,使发泡性片的厚度为1.2mm,将md方向拉伸倍率调整为2.0,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0165]
<实施例14>
[0166]
使牵伸率为1.0,使发泡性片的厚度为1.6mm,将md方向拉伸倍率调整为3.1,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0167]
<实施例15>
[0168]
使牵伸率为1.6,使发泡性片的厚度为1.2mm,将md方向拉伸倍率调整为2.0,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0169]
<实施例16>
[0170]
使牵伸率为1.0,将md方向拉伸倍率调整为2.7,除此以外,按照日本特开2015-187232所记载的实施例6而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0171]
在混合了烯烃系弹性体树脂(dow制,商品名“infuse(注册商标)9107(mfr:1.0g/10分钟)”)33质量份、聚丙烯系树脂(sunoco chemicals制,商品名“tr3020f(mfr:2.1g/10分钟)”)67质量份的基材树脂100质量份中,添加发泡剂(永和化成工业制,商品名:
“ビニホール
(注册商标)ac#r”)6.5质量份、抗氧化剂(basf制,商品名:“irganox(注册商标)1010”)1质量份、交联助剂(和光纯药工业制,80%二乙烯基苯)4质量份,使用亨舍尔混合机进行了混合。在牵伸率1.0、160℃的温度条件下,利用挤出机进行熔融挤出,使用t型模,制作出厚度1.3mm的聚烯烃系树脂片(发泡性片)。
[0172]
将所得的聚烯烃系树脂片在加速电压700kv、电流65ma、照射速度14.4m/min的条件下连续地向一面照射电子射线,获得了交联了的发泡性片。
[0173]
将卷状的交联了的发泡性片在220℃的温度的盐浴上浮起,从上方用红外线加热器加热,将md方向拉伸倍率调整为2.7而使其发泡。用60℃的水冷却,获得了聚烯烃系树脂发泡片。
[0174]
<实施例17>
[0175]
将牵伸率调整为1.0,除此以外,按照日本特开2015-187232所记载的实施例6而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0176]
将md方向拉伸倍率调整为3.1,除此以外,与实施例16同样地操作而制作。
[0177]
<实施例18>
[0178]
将md方向拉伸倍率调整为2.7,除此以外,按照日本特开2015-187232所记载的实施例7而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表2中。
[0179]
关于基材树脂的混配比例,将烯烃系弹性体树脂变更为40质量份,将聚丙烯系树脂变更为60质量份,将牵伸率调整为1.6,除此以外,与实施例16同样地操作而制作。
[0180]
<比较例2,3>
[0181]
使发泡性片的厚度为1.6mm,将md方向拉伸倍率调整为3.1,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0182]
<比较例7>
[0183]
使牵伸率为1.6,使发泡性片的厚度为1.6mm,将md方向拉伸倍率调整为3.1,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0184]
<比较例8>
[0185]
使加速电压为1000kv,使发泡性片的厚度为1.6mm,将md方向拉伸倍率调整为3.1,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0186]
<比较例9>
[0187]
按照日本特开2015-187232所记载的实施例6而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0188]
在混合了烯烃系弹性体树脂(dow制,商品名“infuse(注册商标)9107(mfr:1.0g/10分钟)”)33质量份、聚丙烯系树脂(sunoco chemicals制,商品名“tr3020f(mfr:2.1g/10分钟)”)67质量份的基材树脂100质量份中,添加发泡剂(永和化成工业制,商品名:
“ビニホール
(注册商标)ac#r”)6.5质量份、抗氧化剂(basf制,商品名:“irganox(注册商标)1010”)1质量份、交联助剂(和光纯药工业制,80%二乙烯基苯)4质量份,使用亨舍尔混合机进行了混合。在牵伸率1.6、160℃的温度条件下,利用挤出机进行熔融挤出,使用t型模,制作出厚度1.3mm的聚烯烃系树脂片(发泡性片)。
[0189]
将所得的聚烯烃系树脂片,在加速电压700kv、电流65ma、照射速度14.4m/min的条件下连续地向一面照射电子射线,获得了交联了的发泡性片。
[0190]
将卷状的交联了的发泡性片在220℃的温度的盐浴上浮起,从上方用红外线加热器加热,将md方向拉伸倍率调整为3.1而使其发泡。用60℃的水冷却,获得了聚烯烃系树脂发泡片。
[0191]
将所得的聚烯烃系树脂发泡片的加热收缩率通过日本特开2015-187232所记载的
方法进行了测定,结果,140℃条件为6.9%。
[0192]
<比较例10>
[0193]
按照日本特开2015-187232所记载的实施例7而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0194]
关于基材树脂的混配比例,将烯烃系弹性体树脂变更为40质量份,将聚丙烯系树脂变更为60质量份,除此以外,与比较例9同样地操作而制作。
[0195]
将所得的聚烯烃系树脂发泡片的加热收缩率通过日本特开2015-187232所记载的方法进行了测定,结果,140℃条件为8.3%。
[0196]
<比较例11>
[0197]
按照日本特开2016-155344所记载的实施例4而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0198]
在混合了烯烃系弹性体树脂(三井化学制,商品名
“タフマー
(注册商标)pn-3560”(mfr:6.0g/10分钟))30质量份、聚丙烯系树脂(
プライムポリマー
制,商品名
“プライムポリプロ
(注册商标)j452hp”(mfr:3.5g/10分钟))50质量份、聚乙烯系树脂(日本
ポリエチレン
制,商品名
“ノバテック
(注册商标)lluj960”(mfr:5.0g/10分钟))20质量份的基材树脂100质量份中,添加发泡剂(永和化成工业制,商品名:
“ビニホール
(注册商标)ac#r”)6.7质量份、抗氧化剂(basf制,商品名:“irganox(注册商标)1010”)1.2质量份、交联助剂(和光纯药工业制,55%二乙烯基苯)4.4质量份,使用亨舍尔混合机进行了混合。在牵伸率1.4、170℃的温度条件下,利用挤出机进行熔融挤出,使用t型模,制作出厚度1.5mm的聚烯烃系树脂片(发泡性片)。
[0199]
将所得的聚烯烃系树脂片在加速电压800kv、照射剂量60kgy的条件下连续地向一面照射电子射线,获得了交联了的发泡性片。
[0200]
将卷状的交联了的发泡性片在220℃的温度的盐浴上浮起,从上方用红外线加热器加热,将md方向拉伸倍率调整为3.2而使其发泡。用60℃的水冷却,将发泡表面水洗后,进行干燥而获得了聚烯烃系树脂发泡片。
[0201]
<比较例12>
[0202]
按照日本特开2016-155344所记载的实施例5而制作出发泡体。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0203]
关于基材树脂的混配比例,将聚丙烯系树脂变更为60质量份,将聚乙烯系树脂变更为10质量份,除此以外,与比较例11同样地操作而制作。
[0204]
<比较例13>
[0205]
使牵伸率为1.0,使片厚度为1.8mm,将md方向拉伸倍率调整为3.5,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0206]
<比较例14>
[0207]
使牵伸率为1.6,使片厚度为1.8mm,将md方向拉伸倍率调整为3.5,除此以外,与实施例1同样地操作而制作。将所得的聚烯烃系树脂发泡片的物性示于表3中。
[0208]
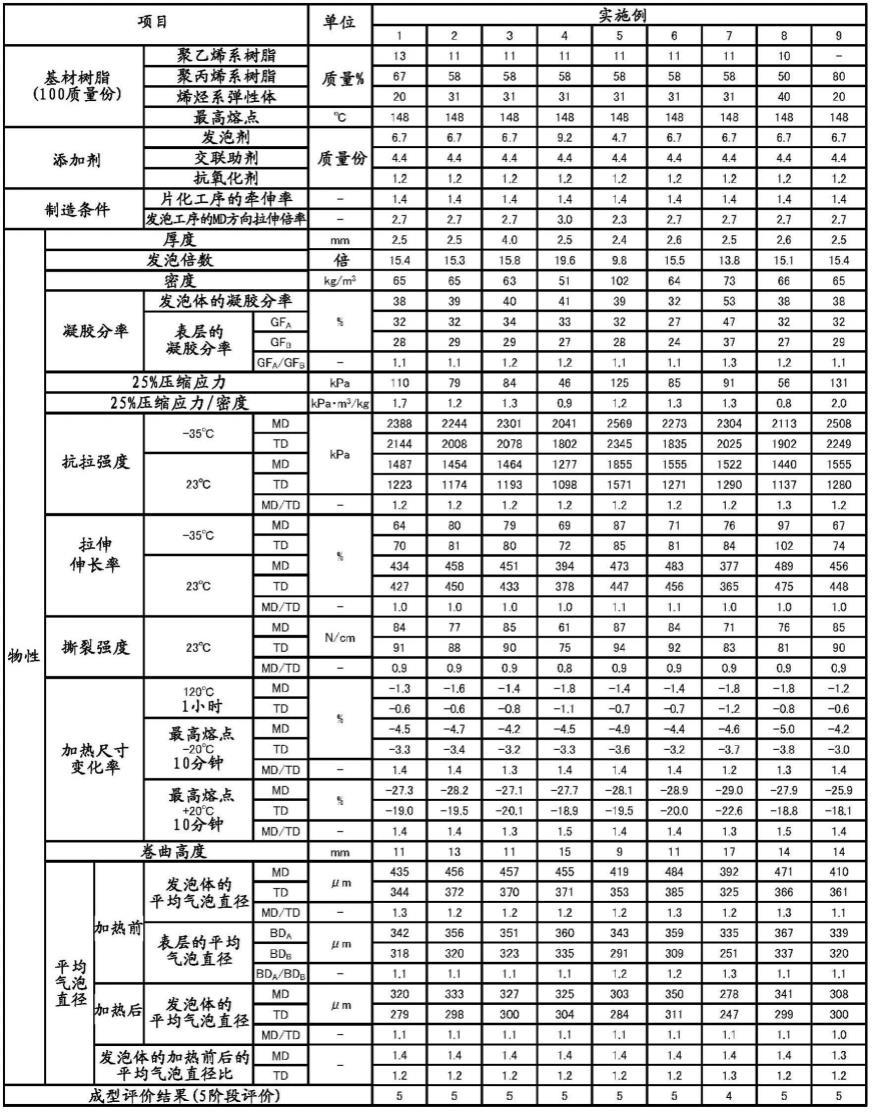
[表1]
[0209]
表1
[0210][0211]
[表2]
[0212]
表2
[0213][0214]
[表3]
[0215]
表3
[0216][0217]
由表1的实施例的结果,“将以聚乙烯系树脂0质量%以上且30质量%以下、聚丙烯系树脂30质量%以上且80质量%以下和聚烯烃系弹性体20质量%以上且40质量%以下混配了的树脂混合物作为基材树脂,在比在dsc测定中作为最高熔融峰的最高熔点高20℃的温度下加热了10分钟时的加热尺寸变化率为-35%以上且0%以下的实施例1~18的聚烯烃系树脂发泡片”确认了柔软性和成型性优异。此外同样地,“将25%压缩应力(kpa)除以密度(kg/m3)而得的值为2.5以下,在dsc测定中作为最高熔融峰的最高熔点+20℃下加热了10分钟时的加热尺寸变化率为-35%以上且0%以下的聚烯烃系树脂发泡片”中,也获得了不发生尺寸缺损的良好的结果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1