染色体邻近实验中的结构变异检测
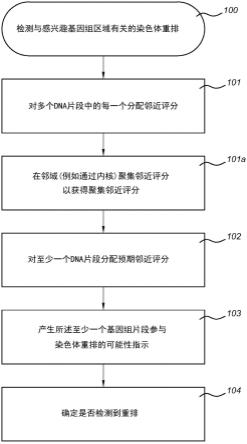
1.本发明涉及分子生物学领域,更具体地涉及dna技术。本发明涉及用于评估感兴趣的基因组区域的dna序列的结构完整性的策略,其在诊断和个性化癌症治疗中具有临床应用。
2.具体提供了一种检测dna读取和感兴趣的基因组区域的染色体重排的方法。将观测邻近评分分配(101)给基因组片段。基于多个基因组片段的观测邻近评分,将预期邻近评分分配(102)给所述多个基因组片段中至少一个基因组片段中的每一个,其中所述预期邻近评分是所述多个基因组片段中至少一个邻近评分的预期值。基于多个基因组片段中的所述至少一个基因组片段的观测邻近评分和多个基因组片段中的所述至少一个基因组片段的预期邻近评分,生成(104)多个基因组片段中的所述至少一个基因组片段参与染色体重排的可能性指示。
背景技术:
3.有一系列技术(3c、4c、5c、hi-c、chia pet、hichip、靶向位点扩增(tla)、capture-c、启动子-capture hic等)(见denker&de laat,genes&development 2016),基于细胞核3d空间中的邻近连接:细胞核内dna片段化和随后的再连接(原位)。在大多数邻近连接测定中,在片段化之前,染色质首先被交联以帮助维持原始3d构象,但也有无交联的原位片段化和邻近连接技术(例如brant等人,mol sys biol 2016)。这些方法在空间邻近(即相互作用)dna片段之间产生连接产物,因此可用于分析细胞核内的染色体折叠。除了邻近连接方法之外,还有其他核邻近方法,例如sprite(通过标签延伸对相互作用的拆分汇集识别)(quinodoz等人,cell 2018),其依赖于交联而不是连接来识别核邻近dna序列。然而,在核(细胞)空间中促成邻近的主要信号是线性邻近。染色体上线性相邻的dna片段在物理上必然是邻近的,这反过来增加了它们在邻近-连接产物或其他核邻近分析中一起被发现的可能性。通常,这种倾向随着染色体上片段对之间线性距离的增加呈指数衰减。
4.这一特征使核邻近方法成为可能,包括邻近连接分析,以灵敏检测导致染色体线性结构变化的染色体重排。例如,进行这种邻近连接测定并分析易位位点附近(靠近两条不同染色体融合的位置)由dna片段形成的连接产物将在两个融合伴侣之间产生非常频繁的连接产物。
5.de laat和grosveld在wo2008084405中公开,可以基于(a)“患病细胞和非患病细胞的dna序列之间的相互作用频率差异”和/或(b)“从低到高相互作用频率的转变”来检测重排。
技术实现要素:
6.在一个方面,本发明提供了一种用于确认染色体断裂点连接的存在,将候选重排伴侣融合到感兴趣的基因组区域内位置的方法,所述方法包括:
7.a、对包含dna的样品进行邻近测定以产生多个邻近连接的产物;
8.b、富集包含基因组片段的邻近连接产物,所述基因组片段包含侧接感兴趣基因组区域5’端的序列,
9.其中所述邻近连接产物进一步包含与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的基因组片段;
10.对所述邻近连接产物进行测序以产生测序读取,
11.将与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的基因组片段序列映射到参考序列;
12.c.富集包含基因组片段的邻近连接产物,所述基因组片段包含侧接感兴趣基因组区域3’端的序列,
13.其中所述邻近连接产物进一步包含与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的基因组片段;
14.对所述邻近连接产物进行测序以产生测序读取,
15.将与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的基因组片段序列映射到参考序列;
16.d.基于所述基因组片段与感兴趣基因组区域或包含侧接感兴趣基因组区域的序列的基因组片段的邻近频率,鉴定至少一个基因组片段作为候选重排伴侣,
17.e、确定与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的候选重排伴侣的基因组片段和与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的候选重排伴侣的基因组片段是重叠的还是线性分离的,
18.其中所述候选重排伴侣基因组片段的线性分离指示所述感兴趣基因组区域内的染色体断裂点连接。
19.优选地,邻近测定是产生多个邻近连接产物的邻近连接测定。
20.优选地,步骤d)包括向基因组的多个基因组片段中的每一个分配(101)观测邻近评分,每个基因组片段的观测邻近评分指示在数据集中存在邻近感兴趣基因组区域且包括对应于基因组片段的序列的至少一个测序读取;
21.基于所述多个基因组片段的观测邻近评分,将预期邻近评分分配(102)给所述多个基因组片段的至少一个基因组片段中的每一个,其中所述预期邻近评分包括所述多个基因组片段中至少一个的邻近评分的预期值;和生成(103)所述多个基因组片段中所述至少一个基因组片段参与染色体重排的可能性指示,该指示基于所述多个基因组片段中所述至少一个基因组片段的观测邻近评分和所述多个基因组片段中所述至少一个基因组片段的预期邻近评分,和将所述基因组片段鉴定为候选重排伴侣。步骤d)的优选实施方式在此进一步描述为plier的实施方式。
22.优选地,步骤b)包括进行寡核苷酸探针杂交或基于引物的扩增,以富集邻近连接产物,所述邻近连接产物包含基因组片段,该基因组片段包含侧接感兴趣基因组区域5’端的序列,和/或步骤c)包括进行寡核苷酸探针杂交或基于引物的扩增,以富集邻近连接产物,所述邻近连接产物包含基因组片段,该基因组片段包含侧接感兴趣基因组区域3’端的序列。
23.优选地,步骤b)包括提供至少一种寡核苷酸探针或引物,其至少部分与侧接感兴趣基因组区域5'区的序列互补,和/或步骤c)包括提供至少一种寡聚核苷酸探针或引物,其
至少部分与侧接感兴趣基因组区域3'区的序列互补。
24.优选地,该方法包括确定将候选重排伴侣融合到感兴趣基因组区域内位置的染色体断裂点连接的位置,所述方法包括:
25.富集包含i)至少部分感兴趣基因组区域和ii)与感兴趣基因组区域邻近的基因组片段的邻近连接产物;
26.对所述邻近连接产物测序并映射染色体断裂点,其中所述映射包括检测i)包含感兴趣的基因组区域的至少第一部分和重排伴侣的基因组片段的邻近连接产物,和ii)包含感兴趣的基因组区域的至少第二部分和重排伴侣的基因组片段的邻近连接产物,其中来自i)和ii)的重排伴侣基因组片段是线性分离的。
27.优选地,该方法包括进行寡核苷酸探针杂交或基于引物的扩增,以富集包含i)至少部分感兴趣基因组区域和ii)与感兴趣基因区域邻近的基因组片段的邻近连接产物。
28.优选地,该方法包括为测序读数的至少一个子集生成矩阵,其中所述矩阵的一个轴表示感兴趣的基因组区域和/或感兴趣基因组区域侧接区域的序列位置,且另一个轴表示候选重排伴侣的序列位置,其中所述矩阵是通过在矩阵上叠加测序读数而产生的,使得所述矩阵内的每个元素表示所识别的邻近连接产物的频率,所述邻近连接产物包含感兴趣基因组区域或侧接感兴趣区域的基因组片段以及来自重排伴侣的基因组片段。优选地,矩阵是蝴蝶图。
29.优选地,该方法包括确定跨越断裂点的基因组区域序列,所述方法包括鉴定包括i)感兴趣基因组区域的断裂点邻近基因组片段和ii)重排伴侣基因组片段的邻近连接产物。
30.优选地,步骤d)包括向基因组的多个基因组片段中的每一个分配(101)观测邻近评分,每个基因组片段的观测邻近评分指示在数据集中存在邻近感兴趣基因组区域且包括对应于基因组片段的序列的至少一个测序读取;
31.基于多个基因组片段的观测邻近评分,将预期邻近评分分配(102)给所述多个基因组片段中至少一个基因组片段中的每一个,其中所述预期邻近评分包括所述多个基因组片段中至少一个邻近评分的预期值;和
32.基于多个基因组片段中的所述至少一个基因组片段的观测邻近评分和多个基因组片段中的所述至少一个基因组片段的预期邻近评分,生成(103)多个基因组片段中的所述至少一个基因组片段参与染色体重排的可能性指示,和将所述基因组片段鉴定为候选重排伴侣。来自步骤d)的优选特征在此进一步描述。例如,在一些实施方式中,将预期邻近评分分配(102)给所述至少一个基因组片段包括:
33.基于多个相关基因组片段的观测邻近评分来确定(303)多个相关邻近评分,其中所述相关基因组片段根据一组选择标准与所述至少一个基因组片段关联;和
34.基于所述多个相关邻近评分确定(304)所述至少一个基因组片段的预期邻近评分。优选地,其中确定(303)多个相关邻近评分包括:
35.生成(401)观测邻近评分的多个置换(permutation),从而鉴定基因组片段中每一个对应的多个置换观测邻近评分,其中生成置换包括交换根据选择标准组彼此相关的随机选择的基因组片段的观测邻近评分。优选地,其中:
36.确定(303)所述至少一个基因组片段的每个相关邻近评分还包括通过聚集置换内
所述至少一个基因组片段的基因组邻域中基因组片段的置换观测邻近评分来聚集(402)一个置换的置换观测邻近评分,以获得每个置换的基因组片段的聚集置换观测邻近评分。
37.还包括聚集(101a)所述至少一个基因组片段的基因组邻域中所述基因组片段的观测邻近评分,以获得所述至少一个基因组片段的聚集的观测邻近评分,
38.其中所述生成(103)所述多个基因组片段中所述至少一个基因组片段是否参与染色体重排的指示是基于所述至少一个基因组片段的聚集的观测邻近评分和所述至少一个基因组片段的预期邻近评分来执行的。优选地,还包括聚集(101a)每个基因组片段的基因组邻域中基因组片段的观测邻近评分,以获得每个基因组片段的聚集的观测邻近评分,其中所述生成(401)置换是基于每个基因组片段的聚集的观测邻近评分,和其中生成(103)所述多个基因组片段中的至少一个基因组片段是否参与染色体重排的指示是基于所述至少一个基因组片段的聚集的观测邻近评分和所述至少一个基因组片段的预期邻近评分来执行的。优选地,对于多个不同规模(501),重复(502)聚集邻近评分(101a),分配(102)预期邻近评分,和生成(103)多个基因组片段中的所述至少一个基因组片段参与染色体重排的可能性指示的步骤,其中在每次重复中(101a’、102’、103’)中,基因组邻域的大小基于所述规模。优选地,确定(304)所述至少一个基因组片段的预期邻近评分包括组合所述至少一个基因组片段的多个相关邻近评分以确定例如平均值和/或标准偏差。优选地,将观测邻近评分分配(101)给多个基因组片段中的每一个包括:
39.向基因组的多个基因组片段分配(201)观测邻近频率,所述观测邻近频率指示在数据集中存在对应基因组片段的至少一个dna读取;和
40.通过组合每个基因组片段的基因组邻域中的观测邻近频率,例如通过对观测邻近频率进行分箱,来计算(202)每个观测邻近评分。优选地,观测邻近频率包括指示与基因组片段对应的dna读取是否存在于数据集中的二进制值,或者指示与数据集中基因组片段对应的dna读取数的值。
41.在一些实施方式中,提供了一种用于确认染色体断裂点连接的存在,将候选重排伴侣融合到感兴趣基因组区域内位置的方法,所述方法包括:
[0042]-确定感兴趣的基因区域;
[0043]-对包含dna的样品进行邻近测定以产生多个邻近连接的产物;
[0044]-富集包含基因组片段的邻近连接产物,所述基因组片段包含侧接感兴趣基因组区域5’端的序列,
[0045]
其中所述邻近连接产物进一步包含与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的基因组片段;
[0046]
对所述邻近连接产物进行测序以产生测序读取,
[0047]
将与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的基因组片段序列映射到参考序列;
[0048]-富集包含基因组片段的邻近连接产物,所述基因组片段包含侧接感兴趣基因组区域3’端的序列,
[0049]
其中所述邻近连接产物进一步包含与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的基因组片段;
[0050]
对所述邻近连接产物进行测序以产生测序读取,
[0051]
将与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的基因组片段序列映射到参考序列;
[0052]-富集包含i)至少部分感兴趣基因组区域和ii)与感兴趣基因组区域邻近的基因组片段的邻近连接产物;
[0053]
对所述邻近连接产物进行测序以产生测序读取,
[0054]
将邻近感兴趣基因组区域的基因组片段序列映射到参考序列;
[0055]-基于所述基因组片段与感兴趣基因组区域或包含侧接感兴趣基因组区域的序列的基因组片段的邻近频率,鉴定至少一个基因组片段作为候选重排伴侣(该步骤的优选实施方式在本文中进一步描述为plier实施方式)
[0056]-确定与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的候选重排伴侣的基因组片段和与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的候选重排伴侣的基因组片段是重叠的还是线性分离的,
[0057]
其中所述候选重排伴侣基因组片段的线性分离指示所述感兴趣基因组区域内的染色体断裂点连接;
[0058]-映射染色体断裂点的位置,包括检测i)包含感兴趣的基因组区域的至少第一部分和重排伴侣的基因组片段的邻近连接产物,和ii)包含感兴趣的基因组区域的至少第二部分和重排伴侣的基因组片段的邻近连接产物,其中来自i)和ii)的重排伴侣基因组片段是线性分离的。
[0059]
在一些实施方式中,提供了一种用于检测染色体断裂点,将候选重排伴侣融合到感兴趣基因组区域内位置的计算机程序产品,所述计算机程序产品包括当被处理器系统执行时,导致处理器系统如下的计算机可读指令:
[0060]-为测序读数的至少一个子集生成矩阵,其中测序读数对应于邻近连接产物的序列,所述产物包括感兴趣基因组区域或侧接感兴趣区域的基因组片段,以及其中至少一个邻近连接产物子集包括候选重排伴侣的基因组片段,
[0061]
其中所述矩阵的一个轴表示感兴趣的基因组区域和/或感兴趣基因组区域侧接区域的序列位置,且另一个轴表示候选重排伴侣的序列位置,其中所述矩阵是通过在矩阵上叠加测序读数而产生的,使得所述矩阵内每个元素表示邻近连接产物的频率,所述邻近连接产物包含感兴趣基因组区域或侧接感兴趣区域的基因组区段以及来自重排伴侣的基因组区段,和
[0062]-检索矩阵以检测轴上的一个或多个坐标,其代表感兴趣的基因组区域和/或感兴趣基因组区域侧接区域的序列位置,其显示来自重排伴侣的基因组区段邻近频率中的转变。
[0063]
在一些实施方式中,处理器系统检索矩阵以检测将矩阵的至少一部分划分为四个象限的一个或多个元素,使得相邻象限之间的频率差异最大化,而相对象限之间的差异最小化。优选地,所述处理器系统:
[0064]-比较鉴定的四个象限和
[0065]-当两个相对象限表现出最小的频率差异而相邻象限表现出最大的频率差异时,将染色体断裂点归类为导致相互重排,或者当一个象限与其他三个象限相比表现出最大频率差异时将染色体断裂点归类为导致非相互重排。
[0066]
优选地,计算机程序产品用于本文公开的任何方法中。
[0067]
能够更准确地检测染色体重排是有利的。为了更好地解决这一问题,提供了一种检测涉及感兴趣基因组区域的染色体重排的方法。该方法在本文中也称为“plier”(基于邻近连接的重排识别),包括:
[0068]
提供从邻近测定(例如,核邻近测定)获得的dna读取的数据集,所述数据集包括表示基因组片段在感兴趣基因组区域附近(例如,细胞核/线性/染色体附近)的dna读取;
[0069]
向基因组的多个基因组片段中的每一个分配观测邻近评分,每个基因组片段的观测邻近评分指示在数据集中存在核邻近感兴趣基因组区域且包括对应于基因组片段的序列的至少一个dna读取;
[0070]
基于多个基因组片段的观测邻近评分,将预期邻近评分分配给所述多个基因组片段中至少一个基因组片段中的每一个,其中所述预期邻近评分包括所述多个基因组片段中至少一个邻近评分的预期值;和
[0071]
基于多个基因组片段中的所述至少一个基因组片段的观测邻近评分和多个基因组片段中的所述至少一个基因组片段的预期邻近评分,生成多个基因组片段中所述至少一个基因组片段参与染色体重排的可能性指示。
[0072]
如本文进一步描述的,该方法和下面描述的优选实施方式可用于基于所述基因组片段与感兴趣的基因组区域或包含侧接感兴趣基因区域的序列的基因组片段的邻近频率来识别作为候选重排伴侣的至少一个基因组片段。
[0073]
预期邻近评分形成特别合适的比较材料,以与观测邻近评分比较,以识别重排。
[0074]
将预期邻近评分分配给所述至少一个基因组片段可包括:基于多个相关基因组片段的观测邻近评分确定多个相关邻近评分,其中相关基因组片段根据一组选择标准与所述至少一个基因组片段关联;和基于多个相关邻近评分确定所述至少一个基因组片段的观测邻近评分。这允许环境特异性预期邻近评分,可能更适合检测染色体重排。
[0075]
确定多个相关邻近评分可包括生成观测邻近评分的多个置换(permutation),从而识别基因组片段中每一个对应的多个置换观测邻近评分,其中生成置换包括交换根据选择标准组彼此相关的随机选择的基因组片段的观测邻近评分。置换可以提供所确定的预期邻近评分的精度改进。
[0076]
确定所述至少一个基因组片段的每个相关邻近评分可包括通过聚集置换内所述至少一个基因组片段的基因组邻域中基因组片段的置换观测邻近评分来聚集一个置换的置换观测邻近评分,以获得每个置换的基因组片段的聚集置换观测邻近评分。这有助于通过减少异常值,使置换邻近评分更真实。此外,或可替代地,它允许确定特定基因组长度规模上的预期邻近评分。
[0077]
该方法可包括聚集所述至少一个基因组片段的基因组邻域中基因组片段的观测邻近评分,以获得所述至少一个基因组片段的聚集的观测邻近评分,其中生成所述多个基因组片段中的至少一个基因组片段是否参与染色体重排的指示是基于所述至少一个基因组片段的聚集的观测邻近评分和所述至少一个基因组片段的预期邻近评分来执行的。这可能有助于增强检测准确性。此外,或可替代地,它允许确定特定基因组长度规模上的预期邻近评分,其可以是用于聚集置换观测邻近评分的相同基因组长度规模。
[0078]
或者,该方法可包括聚集每个基因组片段的基因组邻域中基因组片段的观测邻近
评分,以获得每个基因组片段的聚集的观测邻近评分,和其中所述生成置换是基于每个基因组片段的聚集的观测邻近评分,和其中生成所述多个基因组片段中的至少一个基因组片段是否参与染色体重排的指示是基于所述至少一个基因组片段的聚集的观测邻近评分和所述至少一个基因组片段的预期邻近评分来执行的。这是另一种提高检测准确性和/或确定在特定基因组长度规模上观测和置换邻近评分的方法。
[0079]
可以根据长度规模聚集观测邻近评分,并且可以根据相同的长度规模聚集置换观测邻近评分。这允许确定指示特定长度规模内重排的显著性得分。
[0080]
对于多个不同范围可重复聚集邻近评分,分配预期邻近评分,和生成所述多个基因组片段中的至少一个基因组片段是否参与染色体重排的指示,其中在每次重复内基因组邻域的大小根据范围而定。通过这种方式,可以提供多范围方法,以识别跨多个范围的染色体重排。
[0081]
确定所述至少一个基因组片段的预期邻近评分可包括组合所述至少一个基因组片段的多个相关邻近评分以确定例如平均值和/或标准偏差。这可以提供预期邻近评分值,该值允许为重排检测提供可靠的显著性得分。
[0082]
向多个基因组片段的每一个分配观测邻近评分可包括向基因组的多个基因组片段分配观测邻近频率,所述观测邻近频率指示在数据集中存在对应基因组片段的至少一个dna读取;和通过组合每个基因组片段的基因组邻域中的观测邻近频率,例如通过对观测邻近频率进行分箱,来计算每个观测邻近评分.这可以通过例如平均原始邻近频率数据(例如原始连接频率数据)中的噪声来改善结果。
[0083]
基因组片段的邻近频率可包括指示与基因组片段对应的dna读取是否存在于数据集中的二进制值。这允许例如独立连接片段。
[0084]
基因组片段的邻近频率可包括指示与数据集中基因组片段对应的dna读取数的值。例如,这允许使用非靶向测定。
[0085]
提供dna读取的数据集可包括确定参考基因组中的感兴趣基因组区域;执行邻近测定以产生多个邻近连接/链接片段(也称为邻近连接产物);对邻近链接产物进行测序;将经测序的邻近连接产物映射到参考基因组;选择多个经测序邻近连接产物,所述邻近连接产物包括映射到感兴趣基因组区域的基因组片段;以及检测连接到至少一种所选的经测序邻近连接产物中感兴趣基因组区域的基因组片段。优选地,提供dna读取的数据集可包括确定参考基因组中的感兴趣基因组区域;执行邻近连接测定以产生多个邻近连接片段;对邻近连接片段进行测序;将经测序的邻近连接片段映射到参考基因组;选择多个经测序邻近连接片段,所述邻近连接片段包括映射到感兴趣基因组区域的基因组片段;以及检测连接到至少一种所选的经测序邻近连接片段中感兴趣基因组区域的基因组片段。这些是提供dna读取的合适方法。如本文进一步所述,邻近测定可包括富集包括基因组片段的邻近连接产物,所述基因组片段包括侧接感兴趣基因组区域5’端的序列,以及富集包括基因组片段的邻近连接产物,所述基因组片段包括侧接感兴趣基因组区域3’端的序列。
[0086]
识别多个与基因组片段相关的相关基因组片段的选择标准组可包括以下至少一个:候选相关基因组片段是否在参考基因组中顺式定位于同样包含感兴趣基因组区域的同一染色体;候选相关基因组片段是否在参考基因组中顺式定位于同样包含感兴趣基因组区域的同一染色体的特定部分;以及候选相关基因组片段是否在参考基因组中反式定位于不
包含感兴趣基因组区域的染色体。这些标准可能有助于提高预期邻近评分的质量。
[0087]
识别多个与基因组片段相关的相关基因组片段的选择标准组可包括以下至少一个:候选相关基因组片段是否定位于与感兴趣基因组区域相同或相似的三维核区室的基因组部分;候选相关基因组片段是否定位于与感兴趣的基因组区域具有相同或相似的表观遗传染色质谱的基因组部分;候选相关基因组片段是否定位于与感兴趣的基因组区域具有相似转录活性的基因组部分;候选相关基因组片段是否定位于与感兴趣的基因组区域具有相似复制时间的基因组部分;候选相关基因组片段是否定位于具有作为感兴趣基因组区域实验产生片段的相关密度的基因组部分;以及候选相关基因组片段是否定位于具有相关密度的作为感兴趣的基因组区域的非可映射片段或片段末端的基因组部分。这有助于使预期邻近评分对环境更灵敏。在所有这些例子中,可以基于一组预定的匹配标准来评估“相同或相似”;例如,“成本函数”或“误差函数”对于不太相似的情况较大,对于更相似的情况较小(接近于零)。
[0088]
用于识别多个相关基因组片段的一组选择标准可包括候选相关基因组片段邻近评分具有指示非零dna读取数的值的要求。这可以提高指示重排的显著性得分的质量。
[0089]
生成所述至少一个基因组片段与染色体重排相关的可能性指示可包括:使用一组选择标准来生成所述至少一个基因片段与染色体重组相关可能性的第一指示,所述选择标准排除了候选相关基因组片段邻近评分具有指示非零dna读取数的值的要求;使用一组选择标准产生所述至少一个基因组片段与染色体重排相关的可能性的第二指示,所述选择标准包括候选相关基因组片段邻近评分具有指示非零dna读取数的值的要求;以及基于所述第一指示和所述第二指示生成所述至少一个基因组片段与染色体重排相关的可能性的第三指示。与单独执行任何一种所提出的方法相比,这种组合可以允许产生更可靠的可能性。
[0090]
根据本发明的另一方面,可以提供一种计算机程序产品,其可以存储在无形的计算机可读介质上。计算机程序包括计算机可读指令,当由处理器系统执行时,所述计算机可读指令使处理器系统:
[0091]
向基因组的多个基因组片段中的每一个分配观测邻近评分,基因组片段的观测邻近评分指示在数据集中存在对应于基因组片段的至少一个dna读取,其中数据集包括从邻近测定(例如核邻近测定)获得的dna读取,所述dna读取表示基因组片段在感兴趣基因组区域附近(例如,细胞核/线性/染色体附近);
[0092]
基于多个基因组片段的观测邻近评分,将预期邻近评分分配给所述多个基因组片段中至少一个基因组片段中的每一个,其中所述预期邻近评分是所述多个基因组片段中至少一个邻近评分的预期值;和
[0093]
基于多个基因组片段中的所述至少一个基因组片段的观测邻近评分和多个基因组片段中的所述至少一个基因组片段的预期邻近评分,生成多个基因组片段中所述至少一个基因组片段参与染色体重排的可能性指示。
[0094]
如上所述的方法和计算机程序优选地应用于用于确认染色体断裂点连接的存在的方法中,以便识别候选重排伴侣,如本文所述。
[0095]
本领域技术人员将理解,上述特征可以以任何认为有用的方式组合。此外,关于该方法描述的修改和变化同样可以应用于设备或计算机程序产品。
[0096]
附图简要说明
[0097]
在下文中,将参考附图通过示例来阐明本发明的方面。这些图是图解,可能并非按比例绘制。在整个附图中,类似的项目可以用相同的参考数字标记。
[0098]
图1示出了说明检测染色体重排的方法的流程图。
[0099]
图2示出了说明确定多个dna片段的邻近评分的方法的流程图。
[0100]
图3示出了说明确定至少一个dna片段的预期邻近评分的方法的流程图。
[0101]
图4示出了说明确定特定基因组片段的多个相关邻近评分的方法的流程图。
[0102]
图5示出了说明染色体重排的规模不变检测方法的流程图。
[0103]
图6示出了使用plier实施方式检测染色体重排的说明性示例。a.在包含映射片段(即邻近连接产物)的给定ffpe-tlc数据集中,b.plier最初将参考基因组分割成等间距的基因组间隔,然后c.计算每个间隔的“邻近频率”,该频率定义为由基因组间隔内被至少一个片段(或邻近连接产物)覆盖的片段的数量。d.通过对每个染色体上的邻近频率进行高斯平滑,e.计算观察到的“邻近评分”,以消除最有可能为虚假的邻近频率中非常局部和突然的增加(或减少)。f.通过对整个基因组中观察到的邻近频率进行计算机内改组,然后跨越每个染色体进行高斯平滑,来估计具有相似性质的基因组间隔(例如,跨染色体存在的基因组间隔)的预期(或平均)邻近评分和相应的标准偏差。h.最后,使用其观测邻近评分和相关的预期邻近评分及其标准偏差来计算每个基因组间隔的z-得分。总之,plier客观地检索所捕获的片段浓度显著增加的基因组间隔,并将其视为重排的主要候选。
[0104]
图7示出了用于检测染色体重排的设备的框图。
[0105]
图8a显示了ffpe-tlc工作流程的示意图。(1)通过样品固定,空间上邻近的序列(红色)优先交联。接下来,去除石蜡,并使样品切片通透,以允许酶接触dna。(2)使用nlaiii将dna片段化,然后(3)连接,这导致共定位dna片段的连接。(4)在交联反转和dna纯化之后,(5)对dna进行下一代测序文库制备。(6)使用杂交捕获探针富集感兴趣的序列。(7)制备的文库进行末端配对illumina测序。b.从针对myc、bcl2和bcl6的典型ffpe-tlc实验中获得的片段的全基因组覆盖率。以蓝色显示的是捕获探针靶向的(+/-5mb)基因组间隔处的覆盖率。myc基因的重排区域(绿色)由簇集在grhpr基因(chr9:31mb-42mb)周围的片段浓度识别,以红色表示。c.ffpe-tlc中使用的探针组不仅检索探针互补基因组序列(蓝色)还检测对于myc(粉色)、bcl2(棕色)和bcl6(橙色)所示的侧接序列(即邻近连接产物)的兆碱基。在重排的情况下(在这种情况下为myc-grhpr),相应的捕获探针也获得源自重排伴侣的片段(grhpr,红色)。对于不携带任何重排的区域(例如,棕色的bcl2或橙色的bcl6),情况并非如此,如grhpr基因座所示。
[0106]
图9:a、plier结构变量识别概述。b.对目标基因和plier识别的重排伴侣之间的邻近连接产物(染色体顶部的绿色弧形)的蝴蝶图如何帮助分辨真正靶向重排(断裂点1-3,在探针靶向区域内)和非靶向重排(断裂点4,在探针目标区域外)的示意性解释。在目标基因座内的相互重排中,该基因座应显示一个5'部分(a部分),该部分优先与伴侣基因座的一侧形成邻近连接产物,并与一个3'部分(b部分)分离,该部分优选接触和连接伴侣基因座另一部分。如果探针靶向区域外顺式存在的断裂点(断裂点4),则无法区分目标基因的5'(a)和3'(b)部分。c.蝴蝶图所揭示的三个相互重排的例子,分别涉及myc、bcl2和bcl6。d.重排可以是非相互的,使得只有一个目标基因座的一部分与伴侣融合,例如使用myc、bcl2和bcl6的蝴蝶图所示。e.识别的扩增事件的例子。这些事件从所有目标基因(myc、bcl2和bcl6基
因)捕获的连接产物的数量增加中显而易见。
[0107]
图10:a.circos图显示了本研究中识别的重排伴侣,用于myc(粉色)、bcl2(棕色)和bcl6(橙色)的易位。由多个目标基因发现的伴侣以粗体表示。在我们的研究中发现给定伴侣的频率用括号表示。此外,在每个circos图的圆周上(以浅蓝色突出显示),点表示在我们的研究中发现与每个伴侣重排的目标基因(即粉色点的myc、棕色点的bcl2、橙色点的bcl6)。b.将blc6的不同部分融合到不同基因组伴侣(chr3和chr5)的非相互易位事件的例子。c.涉及igh、myc、bcl2以及chr8和chr10上区域的复杂三向重排的示例,如蝴蝶图和示意图所示。d.bcl6的两个等位基因独立参与重排的例子。e.我们研究中myc基因座中确定的断裂点位置概述。通过映射ffpe-tlc捕获的融合读取,在碱基对分辨率中识别出这样的断裂点。
[0108]
图11:a.稀释样品中plier识别的重排概述。绿色勾表明plier成功识别了易位,基因组中没有任何假阳性调用(calls)。红色叉表明plier未能检测到重排,可能是因为缺乏重排或是因为其他区域的假阳性调用。b.连接产物的可视化以及plier计算的含有bcl2-igh重排的样品f46不同稀释度的富集评分。c.fish对myc中断裂呈阴性的f16和f221的蝴蝶图示。ffpe-tlc显示,它们实际上在同一染色体内存在myc重排。d.fish错过的三个bcl6重排(f38、f40、f49)的蝴蝶图示。在两种情况下(f38、f40),fish未能识别重排,因为具有断裂的细胞百分比低于阈值。e.在f49中,ffpe-tlc显示tbl1xr1基因座1.35mb区域插入到bcl6基因座中。f.f46的bcl6 fish图像显示初始检查时无断裂。事后来看,放大视图(橙色框)显示了一些分裂信号(白色箭头),表明存在易位,如ffpe-tlc检测到的。
[0109]
图12:a.fish、capture-ngs和ffpe-tlc结果的比较显示19个样品中myc、bcl2和bcl6基因中识别的重排。每个圆圈都是一个样品,用于分析特定基因中的重排。实心圆圈表示与fish诊断一致,空心(红色)圆圈表示与fisher诊断不一致。b.capture-ngs的假阳性调用的例子。由于断裂点(红色箭头)周围区域缺少捕获探针,因此缺少ngs读取,对于样品f190无法识别断裂点。ffpe-tlc和plier对sv的识别不依赖于融合读取,正确调用了易位(z评分为82.4)。c.即使断裂点发生在远离被探测区域的地方,ffpe-tlc也能检测易位。每张图从左到右显示了对于两个样品中特定基因的这种能力:bcl2-igh(针对f46和f73显示)、bcl6-igl(针对f37和f45显示)和myc-igh(针对f50和f59显示)。每张图中x轴表示最后一个探针和断裂点位置之间的最小距离。y轴显示plier计算的富集评分。在所有测试的例子中,plier可信地识别易位,即使探针位于距离断裂点50kb处。d.图中显示了本研究中无法在不同映射长度下独特映射到参考序列上的断裂点序列的比例。e.capture-ngs假阳性调用的例子。发现了连接myc基因座和x染色体的断裂点跨越读取,但plier没有发现样品f189的易位峰。使用chrx上的引物的pcr和测序证实了来自chr8的240bp片段的整合,如所示。
[0110]
图13:fish诊断与ffpe-tlc结果的比较。水平fish诊断和垂直ffpe-tlc调用(使用plier)的样品定量概述。注意,“非决定性”fish结果是指携带异常或不均匀数量fish信号的样品。
[0111]
图14:ffpe-tlc样品中读取结构的示意图。ffpe-tlc样品以末端配对模式进行illumina测序。探测的片段(以浅绿色显示)可以仅在一个读取端或在两个读取端上表示。除了这些片段之外,还可以存在邻近连接片段(以蓝色显示)。通过限制位点识别序列(显示为橙色的垂直线)可识别这些片段,该序列将这些片段与被探测片段连接起来。邻近连接片
段可能来自被探测区域周围,或者如果在被探测区域内部或其附近存在重排,则来自重排伴侣附近。如果存在重排,ffpe-tlc读取还可以携带通过探测的(或邻近连接)片段与来自重排伴侣的序列融合而产生的片段(以红色显示)。这种读取可以以碱基对分辨率描述重排事件,并因此提供关于存在的结构变体的更进一步的细节。
[0112]
图15:稍后使用蝴蝶图识别不相关的plier调用示例。a.在样品f209中,当从blc6观察时,plier发现pten基因附近chr10:91mb周围的富集评分显著增加(上图)。然而,当从pten观察时,在bcl6处没有看到倒易峰,但距离bcl6约4.5mb。该观察结果证实,重排没有发生在感兴趣区域内(在这种情况下为bcl6)。b.无关例子的存在可以在最左侧蝴蝶图中描绘的相同例子(即从bcl6看f209)的蝴蝶图示进一步验证。如图所示,看不到覆盖范围的转变(或断裂点)。相反,观察到覆盖率的垂直模式。我们观察到另外两个具有相似特征的例子。从bcl6观察时,f262中出现了一个例子,与f209中已经描述的例子非常相似。另一个例子在f233内,也是从bcl6观察的,但这一次,垂直覆盖率在chr10:104周围有所增加。因此,这三起例子均被视为与plier无关的调用。
[0113]
图16:使用ffpe-tlc中捕获的融合读取在bcl2、bcl6和igh中发现的断裂点概述。
[0114]
ffpe-tlc中的融合读取可以碱基对分辨率映射存在的重排断裂点。该图显示了在我们研究的所有样品中,从bcl2、bcl6和igh myc?观察到的已识别断裂点。
[0115]
图17:稀释度覆盖率对富集评分
[0116]
图18:探针细节
具体实施方式
[0117]
以下将参考附图更详细地描述某些示例性实施方式。提供本说明书和附图中公开的内容,例如详细构造和元件,以帮助全面理解示例性实施方式。因此,显而易见的是,可以在没有那些具体限定内容的情况下实施示范性实施方式。此外,没有详细描述公知的操作或结构,因为它们会用不必要的细节模糊描述。
[0118]
定义
[0119]
在以下描述和实施例中,使用了许多术语。为提供对说明书和权利要求更清楚、一致的理解,包括这些术语的范围,提供以下定义。除非另外定义,本文使用的所有技术和科学术语的意义与本发明所属领域普通技术人员通常所理解的相同。本说明书中提及的所有出版物、专利申请、专利和其他参考文献的公开内容通过引用全部并入本文。
[0120]
实施可用于本发明方法的常规技术的方法对于本领域技术人员来说是显而易见的。分子生物学、生物化学、计算化学、细胞培养、重组dna、生物信息学、基因组学、测序和相关领域中常规技术的实践是本领域技术人员所熟知的,例如在以下参考文献中进行了讨论:sambrook等人,molecular cloning.a laboratory manual,第二版,cold spring harbor laboratory press,cold spring harbor,n.y.,1989;ausubel等人,current protocols in molecular biology,john wiley&sons,new york,1987以及其定期更新,和酶学方法丛书,academic press,san diego。
[0121]
如本文所用,除非文中另有明确说明,单数形式的“一个”、“一种”和“该/所述”包括复数指代形式。例如,如上所述,分离“一个”dna分子的方法包括分离多个分子(例如,10个、100个、1000个、10000个、100000个、数百万或更多个分子)。
[0122]
如本文所用,表述“感兴趣的基因组区域”是指希望评估(至少部分)其结构完整性的生物体染色体的dna序列。例如,被怀疑包含与疾病相关的易位的基因组区域可以被定义为感兴趣的基因组区域。感兴趣的基因组区域可以是单个dna片段、基因、包含基因的基因组位点、染色体的一部分等。
[0123]
在一些实施方式中,感兴趣的基因组区域对应于“拓扑关联域”(tad)。tad由dna-dna相互作用频率定义,其边界是发生相对较少dna-dna相互作用的区域。tad平均为0.8mb,可能含有几个蛋白质编码基因。tad边界通常由是生物体的不同细胞类型共有的,并且富含绝缘子结合蛋白ctcf。tad内基因的表达有一定相关性,因此一些tad倾向于具有活性基因,而另一些tad则倾向于具有抑制基因(参见,例如,dixon等人,nature.2012年5月17日;485(7398):376-380)。
[0124]
如本文所用,术语“基因”是指开放阅读框和与该开放阅读框相关的所有遗传元件。这些遗传元件可能包括内含子、外显子、起始密码子、终止密码子、5'-非翻译区、3'-非翻译区、终止子、增强子位点、沉默子位点、启动子、替代启动子、tata盒和/或caat盒。在原核环境中,“基因”也可能指操纵子,可能包括多个开放阅读框。在一些实施方式中,感兴趣的基因组区域是指在5'非翻译区(5'utr)开始并在3'utr结束的基因序列。用于预测开放阅读框以及上述遗传元件的方法是本领域技术人员公知的。这些方法也称为结构注释,可以利用ejigu和jung(biology 2020,9(9),295;https://doi.org/10.3390/biology9090295)综述的许多不同数据库和计算机算法。
[0125]
如本文所用,表述“开放阅读框”是指起始密码子和终止密码子之间的遗传元件,包括起始密码子与终止密码子。
[0126]
如本文所用,表述“断裂点簇集区”也称为“断裂点成簇区”,是指开放阅读框或基因的子序列,本领域技术人员已知大量患者、生物体或样品中在其中发生或已发生染色体重排。如本领域技术人员所知,一些基因组区域包括几个断裂点簇集区,其可以进一步定义为主要断裂点簇集区和次要断裂点簇集区。
[0127]
如本文所用,术语“等位基因”是指特定基因座上的一种或多种替代形式的基因。在生物体的二倍体细胞中,给定基因的等位基因位于染色体上的特定位置或基因座(多个)。一对同源染色体的每条染色体上都有一个等位基因。因此在二倍体细胞中可能存在两个等位基因,从而存在两个单独(不同的)感兴趣基因组区域。
[0128]
如本文所用,表述“核酸”可指嘧啶和嘌呤碱基的任何多聚物或寡聚物,优选胞嘧啶、胸腺嘧啶和尿嘧啶,以及腺嘌呤和鸟嘌呤(参见albert l.lehninger,principles of biochemistry,第793-800页,worth pub.1982)。本发明涉及任何脱氧核糖核苷酸、核糖核苷酸或肽核酸组分及其任何化学变体,例如这些碱基的甲基化、羟甲基化或糖基化形式等。多聚物或寡聚物的组成可以是异源或同源的,并且可以从天然来源分离,或人工或合成生产。此外,核酸可以是dna或rna或其混合物,并且可以以单链或双链形式永久或瞬时存在,包括同双链体、异双链体和杂交状态。
[0129]
如本文所用,表述“样品dna”是指从生物体或生物体组织或从组织和/或细胞培养物获得的样品,其包含基因组dna。基因组dna编码生物体基因组,这是遗传的生物信息,从生物体的一代传递到下一代。来自生物体的样品dna可从任何类型的生物体获得,例如微生物、病毒、植物、真菌、动物、人类和细菌,或其组合。例如,来自疑似细菌和/或病毒感染的人
类患者的组织样品可以包括人类细胞,但也包括病毒和/或细菌。样品可以包括细胞和/或细胞核。样品dna可以来自患者或对象,其可能面临风险或被怀疑患有特定疾病,例如癌症或任何其他需要调查生物体dna的疾病。
[0130]
如本文所用,表述“交联”是指dna在两个不同的位置发生反应,使得这两个不同位置作为dna链之间的共价键相互连接。两条dna链可以使用uv辐射直接交联,在dna链之间直接形成共价键。两个不同位置之间的连接可以是间接的,通过试剂,例如交联剂分子。第一dna区段可以共价连接到包含两个反应性基团的交联剂分子的第一反应基团,交联剂分子中的第二反应基团可以共价连接至第二dna区段,从而通过交联剂分子间接交联第一和第二dna区段。也可以通过一个以上的分子在两条dna链之间间接形成交联。例如,可以使用的典型交联剂分子是甲醛。甲醛诱导共价蛋白质-蛋白质和dna-蛋白质交联。因此,甲醛可以通过其相关蛋白将不同的dna链彼此交联。例如,甲醛可以与蛋白质和dna反应,通过交联剂分子共价连接蛋白质和dna。因此,可以使用甲醛交联两个dna片段,在第一dna区段和蛋白质之间形成连接,蛋白质可以用连接到第二dna片段的另一甲醛分子形成第二连接,从而形成可以表示成dna1-交联剂-蛋白质-交联剂-dna 2的交联。在任何情况下,应当理解,根据本发明的交联可以包括在物理上彼此接近的dna链之间形成共价连接(直接或间接)。dna链在细胞中可能在物理上彼此接近,因为dna是高度组织化的,同时从序列角度来看,例如相隔例如100kb。只要交联方法与随后的片段化和连接步骤兼容,就可以考虑这样的交联。
[0131]
如本文所用,表述“交联dna样品”是指已进行交联的样品dna。交联样品dna具有这样的效果,即样品内基因组dna的三维状态基本上保持完整。如此,物理上彼此接近的dna链保持在彼此附近。“交联dna样品”可以用福尔马林固定并石蜡包埋:它可以是作为福尔马林固定石蜡包埋(ffpe)材料保存和存储的组织或肿瘤切片或活检。“交联dna样品”可能是ffpe样品或病理学研究常规采集的肿瘤样品。“交联dna样品”也可以是已交联的重构染色质,其中从细胞中分离的基因组dna(例如组织样品或dna样品)进行染色质重构,或以其他方式包装或包覆促进交联的蛋白质或分子,随后进行交联。交联dna的样品包括基因组dna。样品可源自细胞或组织样品。在一些实施方式中,交联dna来自细胞、组织或核样品的交联染色质。虽然在优选实施方式中样品来自人患者,但也可以使用来自其他生物体的dna。
[0132]
如本文所用,表述“逆转交联”包括破坏交联,使得已经交联的dna不再交联,并且适合于后续步骤,例如连接、扩增和/或测序步骤。例如,对已与甲醛交联的样品dna进行蛋白酶k处理将消化样品中存在的蛋白质。由于交联dna通过蛋白质间接连接,蛋白酶处理本身可能会逆转dna之间的交联。保持与dna连接的蛋白质片段可能阻碍随后的测序和/或扩增。因此,逆转dna和蛋白质中氨基酸之间的连接也可能导致“逆转交联”。dna-交联剂-蛋白连接可通过加热步骤逆转,例如在70℃下孵育。由于在交联dna中可能存在大量蛋白质,因此通常需要另外用蛋白酶消化蛋白质。因此,可考虑任何“逆转交联”方法,其中在交联样品中连接的dna链不再连接,并且变得适合于测序和/或扩增。
[0133]
如本文所用,表述“片段化dna”是指当应用于dna(可能是交联dna或不是)时,产生dna“片段”的任何技术。众所周知的dna片段化技术是超声、剪切和/或限制性酶,但也可以考虑其他技术。
[0134]
如本文所用,表述“限制性内切酶”或“限制性酶”可以是识别双链dna分子中特定核苷酸序列(识别位点)的酶,并将在每个识别位点处或附近切割dna分子的两条链,留下钝
端或3
’‑
或5
’‑
突出端。所识别的特定核苷酸序列可决定切割的频率,例如,平均每4096个核苷酸出现一次的6核苷酸的核苷酸序列,而平均每256个核苷酸出现一次的4核苷酸序列则更频繁。
[0135]
如本文所用,表述“连接”涉及单独dna片段的连接。dna片段可以是钝端或可具有相容的突出端(突出粘性末端),使得突出端可彼此杂交。dna片段的连接可以用酶,使用连接酶(即dna连接酶)。然而,也可以使用非酶连接,只要dna片段被连接,即形成共价键。通常,在单独链的羟基和磷酸基团之间形成磷酸二酯键。
[0136]
如本文所用,表述“寡核苷酸引物”或“引物”通常指可引发dna合成的核苷酸链。没有引物,dna聚合酶不能从头合成dna。引物与dna杂交,即形成碱基对。可形成碱基对且彼此互补的核苷酸是例如胞嘧啶和鸟嘌呤、胸腺嘧啶和腺嘌呤、腺嘌呤和尿嘧啶、鸟嘌呤和尿嘧啶。引物和现有dna链之间的互补性不必是100%,即不是引物的所有碱基都需要与现有dna链碱基配对。从与现有dna链杂交的引物3'端使用现有链作为模板(模板引导的dna合成)引入核苷酸。我们可以将用于扩增反应的合成寡核苷酸分子称为“引物”。
[0137]
如本文所用,表述“寡核苷酸探针”或“探针”通常指(修饰的)rna和/或(修饰的)dna核苷酸链,其与连接/链接到细胞核中与感兴趣基因组区域序列相邻的片段的感兴趣基因组区域序列互补,杂交,将其拉下和提取,如例如在capture-c、启动子-capture c、靶向染色质捕获(t2c)、tiled-c和启动子-capture hi-c法中进行的(hughes等人,2014;kolovos等人,2014年;cairns等人,2016;martin等人,2015年;javierre等人,2016年;dao等人,2017年;choy等人,2018年;mifsud等人,2015;montefiori等人,2018;等人,2015,orlando等人,2018,chesi等人,2019年;oudelaar等人,2019)。修饰探针包括例如xgen锁定探针(5
’‑
生物素化寡核苷酸)。
[0138]
如本文所用,术语“杂交”是指通过碱基配对结合两条核酸链。诸如来自探针和引物的核酸序列优选具有与其目标序列至少90%、95%或100%相同的连续序列(例如15-100bp)。如本领域技术人员所知,选择性或特异性杂交取决于例如盐和温度条件。优选使用严格杂交条件,使得探针或引物仅与其靶序列结合。
[0139]
如本文所用,表述“基于引物的扩增”是指多核苷酸扩增反应,即从一条或多条起始序列(即引物)复制的多核苷酸群。合适的引物可以长例如15-30个核苷酸。扩增可指各种扩增反应,包括但不限于聚合酶链式反应(pcr)、线性聚合酶反应、基于核酸序列的扩增、滚环扩增、等温扩增等。合适的基于引物的扩增方法还包括区域特异性提取(rse)(dapprich等人,bmc genomics,2016;17:486)、分子反向探针环化(porreca等人,methods 2007年11月;4(11):931-6)和环介导等温扩增(lamp)(参见例如,notomi等人,nucleic acids res 2000年6月15日;28(12):e63)。
[0140]
如本文所用,表述“测序”是指确定核酸样品(例如dna或rna)中核苷酸(碱基序列)的顺序。许多技术是可用的,如sanger测序和“高通量测序”技术,在本领域中也称为下一代测序,如罗氏、illumina和applied biosystems提供的,或在本领域也称为第三代测序,如david j munroe和timothy j r harris在nature biotechnology 28,426
–
428(2010)中所述,还可使用pacific biosciences和oxford nanopore technologies提供的。这些技术允许在一次运行中从一个样品dna产生多个序列读取。例如,在高通量测序技术的单次运行中,序列读取的数量可以从几百到数十亿。高通量测序技术可根据制造商的说明(如roche、
illumina或applied biosystems提供的)进行。本文设想了长读数和短读数测序方法。该技术可能涉及在进行测序之前制备dna。这种制备可包括将衔接子连接到dna上。衔接子可包括用于区分样品的标识符序列。根据适合或兼容所使用的高通量测序技术的dna的大小,要测序的dna可以进行片段化步骤。“衔接子”是一种短双链寡核苷酸分子,具有有限数量的碱基对,例如长度为约10至约30个碱基对,设计成使得它们可以连接到片段末端。衔接子通常由两个合成寡核苷酸组成,它们具有彼此部分互补的核苷酸序列。这种衔接子可以与基于pcr的富集策略和/或用于邻近连接分子的测序结合使用。
[0141]
如本文所用,表述“测序读取”是指通过核酸测序仪(如大规模平行阵列测序仪(例如illumina或加利福尼亚州太平洋生物科学公司))测序(“读取”)的dna片段。测序读取可包括基因组片段或邻近连接分子的一部分。测序读取可以被映射到参考序列和/或通过例如比对在计算机内组合,以产生连续序列。在一些实施方式中,所述方法产生至少1000、至少5000或至少10000个测序读取。测序读取的数量可以是指与邻近连接分子相对应的测序读取的数目,所述邻近连接分子包括侧接感兴趣的基因组区域5’端的序列;包含侧接感兴趣基因组区域3’端的序列;或包含侧接感兴趣基因组区域的5'端和3'端序列的两个邻近连接分子。测序读取的数量也可以指包含感兴趣基因组区域片段的邻近连接分子。如本领域技术人员所清楚的,如此广泛的测序读取的映射需要使用本领域已知的计算机程序。
[0142]
本文中使用的术语“比对”指基于相同或相似核苷酸的短或长片段的存在来比较两个或多个核苷酸序列。用于比对的方法和计算机程序在本领域中是众所周知的。可用于或改编用于比对的计算机程序是“align 2”,由genentech,inc.编写,于1991年12月10日与用户文档一起提交给美国版权局,华盛顿特区,邮编:20559。
[0143]
如本文所用,表述“参考基因组”(也称为参考组件)是指数字核酸序列数据库,由例如科学家柱状成物种基因集合的代表性示例。由于参考基因组通常是从多个供体dna测序中组装而成,因此参考基因组不能准确地代表任何一个人的一组基因。相反,参考提供了来自每个供体的不同dna序列的单倍体镶嵌。例如,grch37,基因组参考序列合作体人基因组(构建37)来自纽约州水牛城的13名匿名志愿者。参考基因组的其他例子包括grch19和crch38。如本领域技术人员将理解的,参考序列也可用于本文所述的方法中。合适的参考序列包括参考基因组以及来自参考基因组的序列子集。
[0144]
如本文所用,表述“独立连接的dna片段”是指连接到源自给定细胞的给定等位基因的感兴趣基因组区域片段的dna片段。在邻近连接测定中,可在测序之前对独立连接的片段进行pcr扩增,因此可进行多次测序。此外,在一些邻近连接方法中,在交联(任选)、片段化和连接之后获得的邻近连接产物可以进一步裂解,例如为了高效pcr扩增、寡核苷酸诱饵捕获下拉和/或测序,在这种情况下,可以对同一独立连接片段的不同部分进行测序。在独立连接片段对测序数据集贡献多个读取的所有此类情况下,可以执行过滤以生成最佳代表独立连接片段集合的数据集。
[0145]
如本文所用,表述“染色体重排”或“结构变化”是指一组遗传和体细胞遗传畸变,包括染色体缺失、染色体倒置、染色体重复和染色体易位,其中染色体缺失和倒置发生在同一染色体内(顺式),染色体重复发生在同一条染色体内(顺式)或两条或多条不同的染色体之间(反式),或导致一个基因座的染色体外拷贝,而其中易位发生在两条不同染色体之间(反式)。染色体重排还包括外源dna(如转基因和转座子)插入引起的重排。在一些实施方式
中,重排伴侣是外源dna。
[0146]
如本文所用,表述“相互重排”可以指非同源染色体的部分交换,其中没有遗传元件丢失,并且其中一条染色体的遗传元件最终融合到第二条染色体,而第二条的遗传元件最后融合到第一条染色体,并且其中参与重排的每个染色体在每个重排事件中具有一个断裂点。“相互重排”或者还可指作为非同源染色体的部分交换的结果产物,其中没有遗传元件丢失,并且其中一条染色体的遗传元件最终融合到第二条染色体,而第二条的遗传元件最后融合到第一条染色体,并且其中参与重排的每个染色体在每个重排事件中至少具有一个断裂点。相互重排可以是天然或人工过程的结果,并且可以在矩阵中识别,其中所述矩阵的元素表示感兴趣的基因组区域中的基因组片段及其重排伴侣的邻近频率。
[0147]
如本文所用,表述“非相互重排”是指遗传元件从一条染色体转移到另一条非同源染色体,其中第二条染色体的遗传元件没有转移到第一条染色体。“非相互重排”也可以指遗传元件从一条染色体转移到另一条非同源染色体的结果,其中第二条染色体的遗传元件没有转移到第一条染色体。“非相互重排”也可能指外源dna的插入。非相互重排可以是天然或人工过程的结果,并且可以在矩阵中识别,其中所述矩阵的元素表示感兴趣的基因组区域中的基因组片段及其重排伴侣的邻近频率。
[0148]
如本文所用,表述“顺式-染色体”是指根据参考基因组包含感兴趣的基因组区域的染色体。通常,在邻近连接技术中,独立连接片段最可能来自顺式染色体。反过来,源自顺式染色体的独立连接片段更可能是位于与感兴趣的基因组区域线性接近的序列,而不是位于距感兴趣的基因区域更大距离的序列。
[0149]
如本文所用,表述“反式-染色体”是指感兴趣的生物体中非顺式染色体的任何染色体。
[0150]
如本文所用,术语“顺式相互作用”是指源自顺式染色体的遗传元件与目标元件在物理上紧密接近。如本文所用,术语“反式相互作用”是指源自反式染色体的遗传元件与目标元件在物理上紧密接近。
[0151]
如本文所用,dna片段的“连接频率”、“连接的频率“、“相互作用频率”和“邻近频率”的表述可以指该dna片段和感兴趣的基因组区域的连接/链接片段的数量,或者,指该dna片段和感兴趣基因组区域的独立连接/链接片段数量。“连接频率”、“链接”、“相互作用频率”和“邻近频率”可以指dna片段与给定dna区段的顺式和/或反式相互作用的数量,这些dna区段源自dna的实际或理论限制性消化,或可指作为dna片段与给定dna区段的顺式和/或反式相互作用的数量的指示值,所述dna区段源自dna的实际或理论限制性消化。它还可以指在给定基因组区间内源自dna的实际或理论限制性消化的区段的数量,其至少被一连接产物覆盖,或在给定的基因组区间内代表源自dna的实际或理论限制性消化的片段的数量,这些片段至少被一个链接产物所覆盖。通常,在邻近链接/连接技术中,顺式相互作用的相互作用频率高于反式相互作用的相互作用频率。“连接频率”、“链接频率”、“相互作用频率”和“邻近频率”也可以指与连接/链接片段的数量或独立连接/链接的片段数量固有相关的值。例如,代表dna片段连接到感兴趣基因组区域概率的p值也可以被认为是连接频率。例如,可以使用二项式检验来计算这样的p值。频率可以是检测到的相互作用次数的归一化值。这种归一化可以包括对样品之间的差异进行归一化,包括样品质量;以及gc含量、可映射性和限制位点频率的归一化。
[0152]
如本文所用,表述“基因组箱(bin)”或“箱”是指染色体区间,其大小通常在5kb和1mb之间,优选在10kb和200kb之间,可以取代dna片段作为分配连接频率的单位。将连接频率分配给给定箱依赖于聚集该箱内包含的dna片段的连接频率的运算符(求和、均值、中值、最小值、最大值、标准差、三角内核、高斯内核、半高斯内核或任何其他类型的加权和参数化运算符)。
[0153]
如本文所用,片段或箱的“基因组邻域”的表述是指参考基因组中给定片段或箱周围定义的线性染色体区间。片段或箱的基因组邻域可以在10000碱基和5兆碱基之间,优选在200000碱基和3兆碱基之间。基因组邻域也可以基于围绕感兴趣片段或箱的片段数量来定义,其中它通常跨越50-15k的片段。
[0154]
如本文所用,表述“观测聚集连接评分”指根据其自身的连接频率和位于其基因组邻域的片段或箱的连接频率给予每个片段或箱的评分。
[0155]
如本文所用,表述“预期聚集连接评分”是指根据由来自相同实验的连接频率的计算机内置换和聚集建模的背景,给予每个片段或箱的双重评分(即平均值和标准偏差),以表示每个片段或箱的最可能观测聚集连接评分(平均值)以及相应的变化(标准偏差)。
[0156]
如本文所用,表述“相关片段”、“相关箱”、“可比片段”和“可比箱”是指根据某些匹配标准相关的片段或箱。这些匹配标准可以是预先确定的,并且可以取决于手头的实验。例如,给定片段的相关片段可以是源自反式染色体、同一反式染色体、顺式染色体的片段或箱,或其类似长度的片段(或具有片段的箱),或类似交联效率、消化效率、连接效率和/或映射效率的片段,或具有类似表观遗传标志物的片段或箱,或具有相似gc含量或核苷酸组成或保守程度的片段或箱,或位于相同空间核区室中的片段或箱(例如通过hi-c方法确定),或其组合。
[0157]
如本文所用,表述“环境敏感的预期聚集连接评分”指通过置换相关片段或相关箱产生的预期聚集连接评分。
[0158]
如本文所用,表述“显著性得分”是指可以通过将每个片段或箱的观测聚集连接评分与预期聚集连接评分或环境敏感的预期聚集连接评分进行比较来计算的得分。
[0159]
如本文所用,表述“核邻近测定”是指能够识别细胞核中邻近感兴趣基因组区域的dna片段的任何方法。核邻近测定的例子是“邻近连接测定”和不依赖邻近连接的核邻近测定。核邻近也可称为染色体邻近或物理邻近。特别地,邻近是指线性邻近,即沿着顺式染色体的邻近。
[0160]
如本文所用,表述“邻近连接测定”是指依赖于邻近dna片段的连接来识别细胞核中邻近感兴趣基因组区域的dna片段的测定。邻近连接测定在本领域也是已知的,并且可以在本文中用作染色体构象捕获测定,并且包括诸如环状染色体构象捕获或染色体构象捕获与测序(4c)技术组合的方法(simonis等人,2006;van de werken等人,2012)和4c技术的变体(例如umi-4c(schwartzman等人,2016)和mc-4c(allahir等人,2018))、hi-c(lieberman-aiden等人,2009)、原位hi-c(rao等人,2014)和靶向基因座扩增(tla)(de vree等人,2014年)。本文所述的邻近连接方法还可以包括使用互补寡核苷酸探针(由(修饰)rna和/或(修饰)dna核苷酸组成)用于杂交、拉下和富集与核中感兴趣基因组区域序列邻近的片段连接的感兴趣基因组区域序列,例如在capture-c、启动子capture-c和启动子capture hi-c方法中进行的(hughes等人,2014;cairns等人,2016;martin等人,2015;javierre等人,2016
年;dao等人,2017;choy等人,2018;mifsud等人,2015年;montefiori等人,2018年;等人,2015,orlando等人,2018,chesi等人,2019年)。邻近连接方法还包括使用免疫沉淀或其他蛋白质或rna导向策略下拉和富集与携带或与特定蛋白质或rna分子结合的感兴趣基因组区域邻近连接的感兴趣序列的方法,例如chia pet(li等人,2012)和hi-chip(mumbach等人,2017)。邻近连接测定和染色体构象方法的示例见(denker和de laat,2016)。在连接之前,可以在交联和不交联的情况下进行邻近连接测定(brant等人,2016)。
[0161]
也可以在不依赖于将邻近dna片段连接到感兴趣基因组区域的情况下,进行核邻近测定(染色体/物理邻近测定),以鉴定核中与感兴趣的基因组区域邻近的dna片段:不依赖于连接但识别细胞核中邻近感兴趣基因组区域的dna片段的核邻近测定的一个例子是sprite(通过标签延伸对相互作用的拆分汇集识别)(quinodoz等人,2018)。
[0162]
如本文所用,术语“邻近连接产物”是指两个或多个彼此邻近且连接的基因组片段。基因组片段可以直接或间接连接。例如,所述基因组片段可以是交联的,并且可以基于例如条形码或标签(例如sprite)来确定连接。此外,所述基因组片段可以彼此连接(例如作为邻近连接测定的结果)。这种邻近链接产物在本文中称为邻近连接产物。本领域技术人员将理解,除非另有规定,否则本文使用的术语邻近连接产物通常也可包括邻近链接产物。
[0163]
如本文所用,表述“感兴趣的基因组区域的接触概貌”是指在参考基因组上绘制的显示被鉴定为与感兴趣的基因区域核邻近的dna片段的基因组图。
[0164]
如本文所用,表述“染色体断裂点连接”和术语“断裂点”是指染色体或染色体序列上的位置,其中染色体和/或dna产物的两个部分由于天然或人工过程而融合在一起。本公开中特别相关的断裂点连接是那些通常不发生在健康或典型患者、生物体或标本中的断裂点连接。
[0165]
如本文所用,术语“矩阵”是指由两个轴组成的数字、值或表达式表。数字、值或表达式可以由各种元素表示,例如颜色或灰度色调。
[0166]
如本文所用,表述“蝴蝶图”是指显示两个群体变量分布的矩阵。例如,矩阵的一个轴可以表示感兴趣基因组区域和/或感兴趣基因区域侧接区域的序列位置,而另一个轴表示候选重排伴侣的序列位置。
[0167]
实施方式
[0168]
图1示出了检测涉及感兴趣基因组区域的染色体重排的方法100。为此,方法100包含多个步骤来分析dna读取的数据集,该数据集可以从核邻近测定中获得,该数据集中包括代表与感兴趣基因组区域核邻近的基因组片段的dna读取。
[0169]
方法100从步骤101开始,确定多个dna片段中每一个的邻近评分。邻近评分可表示dna片段在基因组上邻近特定感兴趣基因组区域的可能性指示。例如,邻近评分可以与连接/链接到特定感兴趣基因组区域的片段的dna读取的集合相关。更一般地,所述读取是映射到dna片段的多个读取,所述dna片段通过检测方法检测到与感兴趣的遗传区域紧密接近。dna片段的邻近评分表示该dna片段与细胞核内感兴趣区域紧密接近的可能性。例如,邻近评分包括指示读取中该dna片段的读取数的邻近频率。可替换地,邻近评分包括在读取中是否存在该dna片段的至少一个读取的指示。或者,邻近评分包括在读取中存在该dna片段的至少一个读取的可能性指示。例如,可以通过访问包括邻近评分的数据库来确定邻近评分。此外,邻近频率可以经历处理步骤,例如分箱,使得邻近评分与基因组片段的分箱相关。
[0170]
在聚集步骤101a中,可以将步骤101的邻近评分聚集作为另一可选步骤,以获得聚集邻近评分。例如,步骤202的邻近评分可以沿着基因组进行移动平均或加权移动平均。加权移动平均可以通过将基因组的邻近评分与合适内核(例如高斯内核(例如采样高斯内核或离散高斯内核))卷积来实现。这也被称为滑动窗口法,其可替代地涉及例如滑动高斯窗口或内核、半高斯窗口或内核、三角形窗口或内核,矩形窗口或内核或其他类型的窗口或内核。聚集步骤101a的结果可以用作步骤103中dna片段的邻近评分。在省略聚集步骤101a的情况下,例如可以使用步骤202的邻近评分。
[0171]
在步骤102中,确定至少一个dna片段的预期邻近评分。可以基于数据库中其他dna片段的观测邻近评分来计算该预期邻近评分。例如,可以计算数据库中与特定实验和/或染色体相关的所有dna片段的平均值和标准偏差,以确定预期邻近评分。或者,可以对dna片段的随机选择进行平均。又或者,可以确定一组相关dna片段,并且可以仅对那些相关片段的邻近评分进行平均。相关片段可以基于例如它们与感兴趣基因组区域的邻近或基于其他相似性标准来选择。这种相似性标准的示例在本说明书他处公开。
[0172]
在步骤103中,将步骤101中确定的至少一个dna片段的邻近评分与该至少一个dna片段的预期邻近评分进行比较。例如,将dna片段的邻近评分与在步骤102中确定的预期邻近评分进行比较。这得到至少一个dna片段参与染色体重排的可能性指示。例如,该指示可以是显著性得分的形式。在某些实施方式中,在步骤102中确定的标准偏差可以包括在比较中,以确定观测邻近评分相对于预期邻近评分的任何偏差的统计显著性。如果发现明显的偏差,则可认为已检测到染色体重排。统计显著性可以表示为显著性得分。应当理解,可以针对观测邻近评分和预期邻近评分都可用的每个基因组片段计算该显著性得分。
[0173]
在步骤104中,确定是否检测到重排。这可以是布尔决策,即,可评估可得的显著性得分,作为每个基因组片段的是/否决策,或者该决策可以是软决策,包括概率或可能性,或者基因组片段参与与感兴趣基因组区域的重排的确定性。该决策可以基于步骤103中计算的显著性得分。在某些实施方式中,步骤103的显著性得分等于步骤104中的软决策输出。
[0174]
然而,在某些其他实施方式中,在作出决策时考虑更多的输入变量,以生成指示可能重排的增强的显著性得分。例如,可以确定在所映射的目标邻近连接/链接片段的基因组邻域中非可映射实验产生的片段的密度。步骤104中的决策可进一步基于该密度,其中优选地,增强的显著性得分与所映射的目标邻近连接/链接片段的基因组邻域中非可映射实验产生的片段密度成正比。更有甚者,可以确定在所映射的目标邻近连接/链接片段的基因组邻域中可映射实验产生的片段的密度。步骤104中的决策可以进一步基于该密度,其中优选地,增强的显著性得分与给定片段的预期聚集邻近评分成反比。
[0175]
在步骤104检测到可能存在涉及特定感兴趣基因组区域和另一特定基因组片段的基因组重排之后,则可选择地通过重新执行整个程序100,使用另一特定基因组片段作为“感兴趣的特定基因组区域”,进一步验证该重排的存在。如果该程序证实了基因组重排,则更确定重排是真实的。
[0176]
图2示出了如在方法100的步骤101中执行的确定多个dna片段的邻近评分的可能方法。
[0177]
在步骤201中,为多个dna片段中的每一个确定邻近频率。优选地,基因组中的大量连续dna片段用于此,以便于稍后的聚集。例如,dna片段的邻近频率可以是该dna片段的读
取数。根据测定,可能优选执行邻近频率的二值化,例如,如果在读取中发现dna片段,则将邻近频率设置为1,如果在读取中未发现dna片段则将邻近频率设为0。
[0178]
在步骤202中,可以将步骤201的邻近频率组合为可选步骤以生成邻近评分。如果未执行步骤202,则邻近频率本身可以是例如邻近评分。步骤202可以包括例如步骤201的邻近频率分箱。例如,可以定义每个具有多个连续碱基的箱,并且可以在每个箱内组合邻近频率。箱大小可以选择,例如,在5千碱基和1兆碱基之间,优选在10,000碱基和200,000碱基之间。例如,尽管可以选择任何合适大小的箱,但箱可以具有25千碱基的大小。例如,可以通过求和或求平均来组合每个箱内的邻近频率。或者,可以执行二项式测试,例如导致在数据库中的读取中出现箱内基因组片段的可能性。这种二项式测试可能特别适合于二值化邻近频率的情况。在分箱后,所得到的邻近评分可以说与覆盖箱内包含的基因组片段的更大基因组片段有关。
[0179]
应当理解,在某些实施方式中,可以仅执行一个聚集步骤(即步骤202或聚集步骤101a,可能与步骤402结合)或根本不执行聚集步骤。然而,包括两个聚集步骤可能是有利的。此外,在替代实施方式中,可以对步骤202使用内核过滤,并对聚集步骤101a使用分箱。
[0180]
图3示出了实现确定至少一个dna片段的预期邻近评分的步骤102的方法实施方式。例如,分析可能限于一个dna片段,或基因组内的某个区域,或整个染色体。或者,可以对整个基因组进行分析。
[0181]
在步骤303中,为要分析的每个基因组片段生成多个相关的邻近评分。邻近评分可以是从步骤101得到的分数。在这方面,需要注意的是,在组合步骤202中进行分箱的情况下,基因组片段可以被认为是基因组片段的“箱”。
[0182]
在本公开中,相关的邻近评分可以是与正在确定预期邻近评分的基因组片段相关的基因组片段的邻近评分。在这方面,当基因组片段满足某些匹配标准时,它们可能彼此相关。例如,同一染色体上的片段,或基因组上某一距离内的片段,或者已知对某一功能或蛋白质有贡献的片段,或其他可比较的片段可被认为是彼此相关的。本说明书其他地方公开了其他匹配标准。在某些实施方式中,将实验中获得的所有基因组片段设置为相关片段。
[0183]
多个相关的邻近评分可以包括相关基因组片段的所有邻近评分。可选地,为了计算效率,相关邻近评分的集合可以由可用的相关邻近评分的随机选择建立。例如,可以收集1000(或任何其他预定数量的)随机选择的相关基因组片段的邻近评分。
[0184]
在步骤304中,对多个相关邻近评分进行统计计算,从而例如平均值和标准偏差被计算为预期邻近评分。或者例如,可以确定相关邻近评分的中值而不是平均值,或者可以确定方差而不是标准偏差。可以使用其他统计方法来计算预期邻近评分或例如邻近评分的概率密度函数的参数。
[0185]
可以根据需要为每个基因组片段计算该预期邻近评分。
[0186]
图4显示了实现步骤303的方法的实施方式,该步骤303确定与多个相关dna片段相对应的多个相关邻近评分。如上文关于步骤303所观察到的,在步骤101中确定的邻近评分可以用作该方法的起点。
[0187]
在步骤401中,置换相关基因组片段的观测邻近评分。如上所述,当基因组片段满足某些匹配标准时,可以认为它们彼此“相关”。因此在该步骤中,根据匹配标准,第一片段的邻近评分可以与和第一片段相关的第二片段的邻近评分交换。因此,每个邻近评分可以
与另一邻近评分交换。交换的特定基因组片段可以随机选择。为了创建随机置换,每个基因组片段可与另一个随机选择的相关基因组片段交换。或者,可以在随机选择的相关基因组片段对之间进行任意数量(例如,固定数量)的交换。此步骤提供置换邻近评分。
[0188]
在步骤402中,可以聚集步骤401的置换邻近评分。优选地,该聚集步骤涉及与对观测邻近评分执行的聚集步骤101a相同的操作。这样,很容易将聚集的观测邻近评分与预期的聚集邻近评分进行比较。例如,如上在步骤101a所述,可以应用移动平均值或离散高斯内核。此步骤提供聚集的置换邻近评分。
[0189]
在步骤403中,可以在与特定dna片段相关联的集合中收集步骤402的聚集的置换邻近评分,以便稍后可以在步骤304中计算预期邻近评分。或者,可以基于步骤402的聚集的置换邻近评分来更新与特定dna片段相对应的某些统计。如步骤404和405所示,可以收集任何期望的基因组片段的聚集的置换邻近评分。这样,可以检测任何数量的基因组片段的基因组重排/不连续性。在许多情况下,收集所研究基因组上所有基因组片段的聚集的置换邻近评分可能是最有用的。
[0190]
在步骤406中,确定聚集的置换邻近评分的集合是否足够大。例如,该步骤可以通过迭代计数器来实现。该步骤可确保预期邻近评分具有足够的统计相关性。例如,可以执行预定数量的置换;例如1000个置换或100000个置换。
[0191]
如果需要更多的置换来将置换邻近评分的集合扩大到期望的置换数量,则在步骤406中,该过程从步骤401继续。否则,在步骤407完成相关邻近评分的收集。
[0192]
应当理解,在某些实施方式中,不需要在集合中存储置换邻近评分的实际值。相反,通过更新某些参数,可以在一个步骤中组合步骤403和304。例如,如果仅需要估计的邻近评分的均值μ和标准偏差σ,则更新置换邻近评分∑xi之和、置换邻近评分的平方和以及置换邻近评分的数量n就足够了。在步骤403中更新这些参数之后,可以舍弃置换邻近评分的实际值xi。在步骤304中随后可计算的均值如:
[0193][0194]
且标准偏差可计算成:
[0195][0196]
在某些实施方式中,聚集步骤可以在一定长度规模内进行。例如,可以使用观测邻近评分的第二聚集步骤101a和置换邻近评分的聚集步骤402以特定规模将的观测邻近评分与预期邻近评分进行比较。例如,当通过高斯滤波实现聚集步骤时,该规模可以考虑高斯内核滤波的标准偏差。其他类型的滤波可具有类似的规模概念。例如,滑动窗口方法的窗口大小可以根据规模而变化。图1至图4的整个过程可以使用不同的规模执行多次。这可能导致不同规模的不同显著性发现。可以组合不同规模的结果以获得规模不变的结果。例如,从不同规模获得的显著性得分的最大值、最小值或平均值被用作最终规模不变的显著性得分。类似地,在某些实施方式中,第一聚集步骤202可以不同规模执行。例如,在分箱的情况下,可以使用不同的分箱尺寸。
[0197]
在某些实施方式中,可以通过如下处理每个dna片段来执行聚集邻域中观测邻近
评分以获得聚集邻近评分的步骤101a和聚集邻近评分的置换的步骤402。鉴定dna片段的多个相邻dna片段。选择dna片段和相邻dna片段的(观测的或置换的)邻近评分。使用聚集运算符(例如移动平均值,例如加权移动平均值(例如高斯加权移动平均值)或另一类型的运算符)沿基因组组合所选的邻近评分,以产生dna片段的聚集邻近评分。在某些实施方式中,相邻dna片段可以如下识别。可以选择距离测量来识别相邻dna片段。距离测量的第一个例子是基因组距离。在这种情况下,选择在基因组长度规模上接近的dna片段,即,距离dna片段小于一定数量碱基(例如200千碱基或750千碱基)的所有片段都可以是相邻的dna片段。距离测量的第二个例子是基因组中dna片段的数量。在这种情况下,与dna片段最接近的k个dna片段可能是相邻的dna片段。例如k=31或k=51。
[0198]
图5显示了涉及感兴趣基因组区域的染色体重排的这种规模不变检测方法的流程图。在图5中,与图1中步骤相似的步骤编号与图1参考数字相同,并带有撇号。规模不变检测方法包含迭代502,以确定步骤103’中不同规模的显著性得分,其中在步骤501中的每个迭代中设置规模。在步骤104’中,可以使用针对各个规模给出的显著性得分来进行重排的最终确定。
[0199]
更详细地说,该方法从步骤101开始,用例如由测定产生的读取为数据库中的多个dna片段中的每一个分配邻近评分。该步骤可与图1中的步骤101相同。示范性实施方式如图2所示。
[0200]
接下来,在步骤501中,设置规模。例如,规模可以表示为碱基数。然而这不是限制。该规模可以是聚集函数的参数,该聚集函数聚集基因组邻域中dna片段的邻近评分。邻域的宽度可以由规模确定。在聚集函数是高斯内核的情况下,规模可以是用于高斯内核的高斯函数的标准偏差。高斯内核尾部可以任选地在适当的点处被截断。在聚合函数是滑动窗口的情况下,规模可以是滑动窗口宽度。例如,可以为分析选择一组预定的规模,在每个迭代502中选择一个规模。这组规模可以有任意数量的规模。要使用的一组规模(例如标准偏差或窗口宽度)的示例是:{1km、1mb、1000mb}。
[0201]
在步骤101a’中,使用如上所述的所选规模,使用所选规模来聚集邻近评分。通过这种方式,获得聚集邻近评分。上文关于步骤101a概述了用于该聚集步骤的合适方法。
[0202]
在步骤102’中,基于选定的规模确定至少一个dna片段的预期邻近评分。将预期邻近评分分配给所述至少一个dna片段。对于dna片段的特定子集,例如基因组区域,可以将预期邻近评分分配给一个dna片段,或者分配给整个染色体或整个基因组的dna片段。例如,如上文图3和图4所公开的,可以实现计算预期邻近评分的方法。在步骤402中,可以使用所选择的规模来聚集邻近评分的置换。例如,可以使用与步骤101a’中相同的聚集算法和聚集参数。
[0203]
在步骤103’中,使用根据步骤101a’的规模的聚集邻近评分和根据步骤102’的规模的预期邻近评分来确定所述至少一个基因组片段参与染色体重排的可能性指示,例如显著性得分。这样,对于每个选定的规模,可以获得染色体重排可能性的不同指示。
[0204]
在步骤502中,验证是否应用了所有期望的规模。如果更多的规模需要计算,则从步骤501重复该过程,其中选择另一个规模。例如,迭代该过程,直到已经选择了预定规模组的所有规模。
[0205]
如果已经对所有期望的规模执行了该过程,则该过程在步骤104’中进行,以基于
在步骤103’中针对所有选定规模确定的指示(显著性得分)来确定是否检测到重排。不同规模的指示(显著性得分)可以多种可能方式之一组合,例如可以确定至少一个dna片段的可得显著性得分的最大值、平均值、中值或最小值。此后可以任选应用阈值以获得二进制确定。然后结束过程。
[0206]
应当理解,上文参考图1至图5描述的方法可以作为计算机程序或适当编程的计算机系统来实现。通过邻近测定创建的数据集可以用作计算机程序输入,并且输出可以是检测到重排的指示。
[0207]
在本文全文中可以理解,连接频率是邻近频率的示例,而连接评分是邻近评分的示例。尽管使用连接频率和连接评分作为示例在整个文档中示出和解释了几种技术,但是应当理解,通常可以使用任何邻近频率和/或邻近评分来执行本文公开的技术。例如,可以使用不依赖于“邻近连接”的核邻近测定,例如sprite方法,来识别感兴趣基因组区域附近的dna片段。因此,在本公开中,术语连接和邻近可以互换使用。具体地,术语连接频率和邻近频率可以互换使用。类似地,术语连接评分和邻近评分可以互换使用。
[0208]
图6显示了应用本文所述方法的说明性示例。作为示例,邻近频率可以作为4c分布或另一种测定技术获得。这样的测定可产生邻近连接数据集。图6显示了沿染色体(部分显示在横轴上)观察到的dna片段的邻近频率(纵轴)的图600。覆盖染色体一小部分的图600的细节显示在图601中。使用具有例如25千碱基宽度的箱来对该分布分箱,以获得观测邻近评分的评分分布。评分分布的细节示于图602中,满分分布示于图603中。在该实施例中,使用高斯内核605来聚集评分分布603,以获得观测聚集邻近评分的聚集或平滑的评分分布,如图606所示。评分分布603被置换以获得随机置换分布604,其也使用高斯内核605平滑。置换和平滑重复n次,其中n是整数,例如1000。如图607所示,从所有这些置换的平滑分布中,导出预期聚集邻近评分的预期分布。例如通过减法(或例如通过平方差)将平滑分布606与预期分布607进行比较,以获得如图608所示的差异分布。还从置换的平滑分布和/或期望分布导出显著性阈值609。可选地,重要性阈值609可以被设置为可配置值。在比较分布608超过显著性阈值609的片段处,如在片段610处所指示的,可以触发对可能重排的指示。
[0209]
图7示出了用于检测染色体重排的设备的框图。该装置可以实现为配置成执行本文公开的任何方法的计算机系统。例如,在获得dna读取后的步骤可以由设备700执行。特别是,检测染色体重排所需的计算步骤可以由该设备执行。例如,设备700可以包括可以执行指令的处理器701。处理器701可以包括配置为协同工作的多个(子)处理器。设备700还可以包括存储器702,存储器702可以是任何数据存储装置,例如闪存或随机存取存储器,或两者。存储器702可以包括非瞬时计算机可读介质。存储器702可以存储指令,使得处理器701在执行指令时执行本文阐述的方法。这些指令可以共同形成计算机程序。计算机程序可替代地存储在单独的非瞬时计算机可读介质上,例如光盘。此外,存储器702可以被配置为存储与测定有关的数据,例如具有dna读取的数据库。诸如dna读取的数据可以经由收发器703接收,收发器703可以是例如通用串行总线(usb)或无线通信设备。此外,该方法的结果,例如指示任何重排的显著性得分,可以通过收发器703输出。外设可以通过收发器703连接。可任选地,设备700包括用户接口组件(未示出),例如显示器和/或用户输入设备,例如鼠标、键盘或触摸板。这样的用户接口组件还可经由收发器703连接。此外,这样的用户接口组件可用于控制设备的操作和/或输出计算结果。例如,收发器703还可以与外部存储器通信。最
后,设备700可替代地实现为分布式计算机系统,其在云服务器上执行计算或数据存储的一部分,在客户端设备上执行另一部分。
[0210]
在某些实施方式中,可以使用称为邻近连接测定的核邻近测定。此外,可以考虑(交联的)dna样品内和样品之间的技术和生物学偏差以及变化,以通过计算识别在感兴趣的基因组区域中发生的结构变化。
[0211]
在某些实施方式中,鉴定在感兴趣基因组区域中发生的结构变化的方法可以包括以下步骤:
[0212]-执行邻近连接测定以产生与感兴趣基因组区域核邻近的独立连接片段的数据集。
[0213]-使用数据集为每个片段分配观测的聚集连接评分。
[0214]-使用相同的数据集计算每个片段的环境敏感的预期聚集连接评分。
[0215]-在不同的染色体长度规模上比较片段观测的相对于环境敏感的预期聚集连接评分,并识别与环境敏感的预期聚集连接评分相比具有显著增加的聚集连接评分的每个染色体长度规模片段。
[0216]
在某些实施方式中,使用不依赖于“邻近连接”的核邻近测定,例如“sprite”方法,识别邻近感兴趣基因组区域的dna片段,并考虑(交联)dna样品内和样品间的技术和生物学偏差和变化,以通过计算识别感兴趣基因组区域中发生的结构变化,包括以下步骤:
[0217]-执行邻近连接测定以产生与感兴趣基因组区域核邻近的dna片段的数据集。
[0218]-使用数据集为每个片段分配观测的聚集邻近评分。
[0219]-使用相同的数据集计算每个片段的环境敏感的预期聚集邻近评分。
[0220]-在不同的染色体长度规模上比较片段观测的相对于环境敏感的预期聚集邻近评分,并识别具有每个具有显著增加的聚集邻近评分的染色体长度规模片段。
[0221]
本文所公开的技术基于期望更准确检测染色体重排的实现。这主要是因为在两个给定样品(例如,患病细胞和健康细胞)的比较中,可以检测到邻近连接产物之间的许多差异,这些差异不是由实际结构变化引起的。此外,在任何邻近连接数据集中可以看到的从低到高相互作用频率的许多转变都不是由结构变化引起的。因此,本发明的一个方面是弥补这些缺点,识别基因组中的基因组结构变化,同时考虑在同一数据集中观察到的内在技术偏差。
[0222]
易位(染色体重排)是不同形式癌症的基础(schram等人,2017)。它们可能导致癌基因的过度表达或产生具有失调表达或激酶活性的融合蛋白。易位的分子分型通常在临床上用于诊断(肿瘤分类)、预后,也越来越多地用于治疗决策。例如,携带蛋白激酶基因alk和ros1易位的非小细胞肺癌(nsclc)可被fda批准的蛋白激酶抑制剂靶向(kwak等人,2010;shaw等人,2014),而ret的强效抑制剂是治疗ret易位患者的有前途的精准药物(plenker等人,2017)。因此,nsclc肿瘤的分子分型(pispia等人,2017)对于选择最佳治疗方法非常有用,并且在荷兰对于iv期(转移性)肺癌是强制的(每年1000例)。此外,还对每年约1500名被诊断为弥散性大b细胞淋巴瘤(dlbcl)的患者以及荷兰每年约700名患有各种形式肉瘤的患者中的许多进行易位分析。
[0223]
几十年来,常规临床程序是将手术切除的肿瘤活检保存为福尔马林固定石蜡包埋(ffpe)标本。然而,ffpe样品中的dna或rna重排检测受到影响,因为dna和rna是交联且片段
化的。存在用于重排检测的基于rna和dna的pcr策略,但很复杂。首先,复现性重排基因的断裂点位置和重排伴侣在患者之间往往不同,这使得设计检测所有可能重排的pcr引物集变得困难。新的融合伴侣经常被遗漏,在这种情况下,当获得阴性结果时,无法形成关于重排的结论性意见。一些基于rna的pcr策略,如archer fusionplex,对于重排伴侣是不可知的,但在异质性肿瘤活检中未发现重排并不排除其存在。此外,ffpe样品中的rna可能太少,或者rna质量太低,不利于随后的cdna pcr产物分析。最后,不产生融合但导致本来未改变的致癌基因上调的所谓位置效应重排,根据定义,在rna水平上是无法检测到的。
[0224]
由于这些原因,荧光原位杂交(fish)通常仍然是检测ffpe活检中融合的首选诊断方法。然而,fish是劳动密集型的,仅提供部分信息,并不总是结论性的。每个基因都需要在独立fish实验中单独测试。如果感兴趣的基因与不同染色体伴侣混杂重排(这可能是经常发生的情况),则使用分离fish(或分裂fish)。分裂-fish需要在靶基因的每一侧杂交不同颜色标记的探针:如果它们分离(“分裂”),即如果它们在给定数量的细胞中分离的距离大于预期,则该基因被认为参与易位,但重排伴侣仍不清楚。此外,由于样品质量和肿瘤负荷,fish可能给出不明确的结果。因此,迫切需要一种强大的、简单的、一体化检测方法,能够同时检测所有感兴趣基因的重排,而不考虑其断裂点位置和易位伴侣。可以使用本文公开的重排检测方法使这种测定成为可能。
[0225]
dna样品或交联dna样品中的重排检测方法优选满足以下标准中的任何一个或多个,理想地全部:
[0226]
(1)一种能够同时监测与给定疾病相关的所有基因重排的一体化方法,
[0227]
(2)一种对于确切断裂点位置和重排伴侣不可知的,因此能够找到已知的和新的易位伴侣的方法,
[0228]
(3)一种足够灵敏的方法,能够在小的(例如,小于5%)细胞亚群中检测到重排,以及
[0229]
(4)提供对重排无偏检测的方法。
[0230]
核邻近测定,例如邻近连接测定,可能满足前三个标准,正如4c技术首次所示。4c技术最初由发明人开发,用于研究基因组的三维折叠(simonis等人,2006)。该方法是3c技术的变体(dekker等人,2002年),允许对与所选感兴趣基因组位点(“视点序列”)紧密接近的所有染色体片段进行无偏全基因组映射。该技术涉及甲醛介导的细胞固定,这导致每个细胞核内物理邻近的dna序列之间的交联。随后用限制性酶消化交联的dna,并在有利于交联dna片段之间邻近连接的条件下重新连接。因此,3c策略在最初在核空间紧密靠近的dna序列之间产生连接产物。在4c技术中,使用视点特异性引物对环状连接产物进行反向pcr,这导致其捕获的连接伴侣的扩增;这些可能随后被illumina测序并映射到基因组以揭示视点的接触分布。
[0231]
正如从聚合物物理学中所预期的,无论3d构象如何,绝大多数4c捕获的片段总是源自与线性染色体模板上的视点紧邻的序列。基于这一认识,发明人在过去假设并证明4c技术非常适合检测染色体重排,包括易位,因为这种染色体畸变会干扰线性染色体骨架(simonis等人,2009年;homminga等人,2011年)。因此,当4c视点位于重排断裂点附近时,它将基于感兴趣基因组区域的改变的接触分布来识别重排和重排伴侣(simonis等人,2009)。尽管当视点和断裂点更靠近时,测定的灵敏度(即其检测小细胞亚群中易位的能力)增加:
对于距离断裂点100kb以内的视点,即使它们存在于少于5%的细胞中,也可以容易地发现易位(simonis等人,nat methods 2009,以及未发表的数据)。后者对于致癌诊断至关重要,因为癌症活检通常是健康和不同克隆癌细胞群体的混合物。总之,4c提供了一种灵敏方法来研究候选基因(例如,人们希望在临床上监测重排的基因)是否参与重排并识别其重排伴侣。已发表的4c的另一个优点(simonis等人,2009)是4c pcr反应可以很容易地多重化,这意味着该测定可以同时监测每个患者样品中的多个基因的重排。
[0232]
除了4c技术,我们现在知道,还有许多其他基于相同原理的邻近连接方法也可以识别具有感兴趣的基因组区域的染色体重排。此类方法的例子是靶向基因座扩增(tla)、capture-c或capture-hic方法、hi-c和原位hi-c、chia-pet和hi-chip。原则上,所有进行邻近连接以识别细胞核中邻近感兴趣基因组区域的dna片段的方法都能够检测染色体重排和易位。
[0233]
邻近连接方法可用于鉴定染色体重排。旨在基于邻近连接方法识别结构变化的现有技术方法通常依赖于对感兴趣的基因组区域的接触分布的目视检查,以在与测试样品(例如来自疾病患者的样品)中与感兴趣基因组区域邻近连接的dna片段的基因组聚类的其他地方(或不聚类)处发现与对照样品(例如,来自健康个体的样品)的同一基因组基因座处所见邻近连接dna片段聚类明显不同的聚类。在对感兴趣的基因组区域的接触分布进行这种目视检查时发现的易位和其他染色体重排的例子见(simonis等人,2009;de vree等人,2014;harewood等人,2017和wo2008084405)。在其他当前实验设计中,将从疾病(例如癌症)细胞产生的测试样品中获得的核邻近数据集与从正常(健康)细胞中产生的对照核邻近数据集进行计算比较,以识别指示染色体重排的核邻近dna片段的异常基因组分布(d
í
az等人,2018)。dixon等人2018通过组合从9个核型正常细胞系产生的核邻近数据集,利用广泛对照数据集来估计预期的染色体间相互作用频率,该频率解释了源自染色体末端或小染色体的片段的相互作用升高。这种测试样品相对对照样品校正方法的缺点在于,它不能解释在核邻近测定(如邻近连接测定)中容易发生的样品特异性偏差。例如,所研究样品的纯度、交联能力、片段化效率和(在邻近连接测定中)连接效率可对与感兴趣基因组区域3d邻近的片段在所产生的核邻近数据集中的表现产生实质性影响。因此,纠正这些隐藏的实验特异性偏差是利用核邻近技术评估易感基因座结构完整性,从而将这些方法用于临床应用的主要障碍。
[0234]
因此,当前的发明人设计了通过考虑数据集特异性技术和实验偏差来识别感兴趣区域中的结构变化的策略。这些策略可包括构建背景模型,该背景模型是从研究中的邻近连接数据集(例如,从患者肿瘤获得的测试样品)计算的,然后利用背景模型来评估跨越相同测试样品基因组的连接dna片段聚类的显著性。在该数据内在分析程序中,可能不需要使用对照样品数据集。
[0235]
发明人已经认识到,涉及感兴趣区域的结构变化(例如染色体重排或易位)的片段将显示出比偶然预期更高数量的独立连接的dna片段。
[0236]
基于上述前提,可以通过本文公开的方法、装置和计算机程序技术来评估感兴趣基因组区域在染色体重排中的参与。
[0237]
在某些实施方式中,可通过以下方式评估感兴趣基因组区域在染色体重排中的参与:
[0238]
a.执行邻近连接测定,其创建具有感兴趣基因组区域的独立连接的dna片段的数据集(本文也称为邻近连接/链接产物)。
[0239]
b.聚集(例如通过求和)每个片段的基因组邻域中的连接频率,为每个片段分配“观测聚集连接评分”。
[0240]
c.用另一个随机选择的dna片段置换(交换)每个dna片段(包括观测连接频率等于零的dna片段)的连接频率。
[0241]
d.聚集每个片段及其相邻片段的置换连接频率,以计算每个片段的随机化聚集连接评分。
[0242]
e.多次重复步骤c-d(通常n=1000),以形成数据集中每个片段的“预期聚集连接评分”。
[0243]
f.可选地,将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。这些片段可以位于距离感兴趣的基因组区域最大延伸10mb的染色体区间中。该步骤f有效排除了侧接感兴趣基因组区域的基因组区域的观测聚集连接评分,其可能具有高的显著性得分,这不是因为参与重排,而是由于与未重排的基因组中的感兴趣区域线性相邻。
[0244]
g.将每个dna片段的观测聚集连接评分与预期聚集连接评分进行比较,以识别具有高显著性的dna片段(即,观测聚集连接评分明显大于预期聚集连接评分)。
[0245]
在某些实施方式中,提供了一种方法来评估感兴趣基因组区域在顺式染色体重排(例如,染色体内缺失、反转或插入)中的参与,并且使用环境敏感的预期聚集连接评分来解释源自顺式与反式染色体的片段的预期连接频率之间的差异,通过以下:
[0246]
a.执行邻近连接测定,其创建具有感兴趣基因组区域的独立连接的dna片段的数据集(本文也称为邻近连接/链接产物)。
[0247]
b.聚集驻留在数据集中每个片段附近的片段的连接频率,以形成每个片段的观测“聚集连接评分”。
[0248]
c.用源自顺式染色体的另一个随机选择片段置换源自顺式染色体的每个片段(包括与观测连接频率等于零的dna片段为顺式的dna片段)的连接频率。
[0249]
d.聚集源自顺式染色体的每个片段及其相邻片段的置换连接频率,以计算源自顺式染色体的每个片段的随机化聚集连接评分。
[0250]
e.多次重复步骤b-d(通常n=1000),以形成数据集中每个片段的预期聚集连接评分。
[0251]
f.可选地,将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。
[0252]
g.将源自顺式染色体的每个片段的观测聚集连接评分与预期聚集连接评分进行比较,以识别含有感兴趣基因组区域的顺式染色体中具有高显著性的片段(即,具有显著增加的观测聚集连接评分)。
[0253]
在其它实施方式中,提供了一种方法来评估基因组区域在染色体间重排(例如,染色体之间易位)中的参与,并且使用环境敏感的预期聚集连接评分来解释源自顺式与反式染色体的片段的预期连接频率之间的差异,通过以下:
[0254]
a.执行邻近连接测定,其创建具有感兴趣基因组区域的独立连接的dna片段的数据集(本文也称为邻近连接/链接产物)。
[0255]
b.聚集驻留在数据集中每个片段附近的片段的连接频率,以形成每个片段的观测
聚集连接评分。
[0256]
c.用源自反式染色体的另一个随机选择片段置换源自反式染色体的每个片段(包括与观测连接频率等于零的dna片段为反式的dna片段)的连接频率。
[0257]
d.聚集源自相同反式染色体的每个片段及其相邻片段的置换连接频率,以计算源自反式染色体的每个片段的随机化聚集连接评分。
[0258]
e.多次重复步骤b-d(通常n=1000),以形成数据集中每个反式dna片段的预期聚集连接评分。
[0259]
f.源自反式染色体的每个片段的观测聚集连接评分与预期聚集连接评分进行比较,以识别反式染色体中具有高显著性的片段(即,具有显著增加的观测聚集连接评分)
[0260]
相邻dna片段的邻近频率的聚集可以包括求和、滚动平均值、滚动中值、最小值、最大值、标准差、三角内核、高斯内核、半高斯内核、或任何其他类型的加权和、或任何其它聚合方法,例如基因组中特定dna片段周围的dna片段窗口内的平方频率值的均值。
[0261]
染色体扩增通常可以在扩增的染色体区段上显示相对均一的邻近频率。然而,重排伴侣通常在伴侣融合到感兴趣基因组区域的断裂点附近具有最高邻近频率。此外,对于离断裂点更远的片段,这种重排伴侣通常可以显示更小的邻近频率。
[0262]
在某些实施方式中,染色体扩增可通过仅在连接到感兴趣基因组区域的片段之间置换邻近频率(例如,在步骤c或步骤401中)而与重排伴侣区分。也就是说,当计算预期聚集邻近评分时,仅置换邻近频率高于零的dna片段。
[0263]
在某些实施方式中,执行本文所公开的几种不同计算方法,以检测染色体重排。为了提高检测准确性,可以组合这些不同计算方法的结果。例如,可以通过使用包括观测邻近频率等于零的dna片段的dna片段置换,或者仅使用非零观测邻近频率的dna片段置换来计算预期聚集邻近频率。然而,也可以使用这两种方法计算两种形式的预期聚集邻近频率,并确定与两种预期聚集邻近频率的任何偏差的显著性,并组合两种方法的结果。例如,只有当两种方法都导致显著的偏差时,才能决定染色体重排。或者,可以通过两种方法确定染色体重排的可能性,并且可以通过组合不同应用方法的可能性来确定染色体重排最终可能性。例如,如上所述,当检测染色体间重排时,可以执行这种组合方法。
[0264]
在某些实施方式中,沿着基因组的dna片段可以分箱,从而对于紧密相关的dna片段的分箱而不是对于每个dna片段单独检测邻近频率。在这种情况下,置换可以是箱的置换,而不是单个dna片段的置换。
[0265]
在某些实施方式中,可以通过将每个dna片段或箱的观测聚集邻近频率与考虑到实验中考虑的所有dna片段或箱内的预期聚集邻近频率进行比较来计算dna片段或箱的观测聚集邻近频率的显著性得分。这种过程可以帮助减少假阳性调用的数量。
[0266]
在某些实施方式中,预期的聚集邻近评分可以是环境敏感的。例如,根据某些标准,dna片段的邻近频率的置换可限于相关dna片段(或箱)之间的交换。“相关片段”和“相关箱”可以例如是源自相同反式染色体的片段或箱,或是源自与感兴趣基因组区域相距一定线性距离的顺式染色体片段的片段或箱,或者是长度相似的片段(或具有片段的箱),或者是具有类似交联、消化、连接和/映射图效率的片段(或具有片段的箱),或者是来自染色体区段的具有相似交联、消化连接和/或映射效率的片段(或者具有片段的箱),或是具有相似表观遗传谱或相似转录活性或相似复制时间(在所研究的细胞类型中)的来自染色体区段
的片段或箱,或者是驻留在同一空间核区室(例如通过hi-c方法确定的a和b区室)中的片段或箱,或其组合。在这些标准中,例如,通过设置交换的两个dna片段(或箱)中相关数量的值之间的最大差异,可以实现“相似”。
[0267]
在某些实施方式中,考虑不同的基因组长度规模以识别涉及感兴趣基因组区域的染色体重排,例如通过考虑邻域聚集的多种大小。例如,该分析可以计算大小为200kb、750kb和3mb的基因组邻域中三种不同基因组长度规模的显著性得分。例如,聚集可以涉及平均n个最近dna片段的邻近频率,其中n是对应于基因组长度规模的整数。或者,通过应用内核,聚集可以涉及相邻dna片段的邻近频率的加权和。例如,内核可以对应于具有标准偏差的高斯分布,其中标准偏差对应于基因组长度规模。类似地,可以使用其他参数化内核,其中内核的参数可以对应于基因组长度规模。
[0268]
在某些实施方式中,可以组合针对基因组邻域的多个不同长度规模计算的显著性得分,以产生“规模不变”的显著性得分。显著性得分组合的典型运算符是最小值和平均值,但也可以使用其他运算符。
[0269]
在某些实施方式中,可以通过使用说明基因组(n)中片段总数的二项式检验,以及dna片段具有映射到它的至少一个读取的机会(其中m是数据集中具有映射到它的至少一个读取的dna片段的总数),校正邻近频率中具有在离散数据集中每个dna片段邻域中映射到它的至少一个读取(k)的dna片段的密度。然后,所得的p值被视为每个片段的邻近频率(见等式1)。将附近片段的邻近频率组合成聚集邻近评分。
[0270][0271]
在某些实施方式中,通过使用两个独立的二项式测试,可以针对顺式与反式染色体中片段的预期邻近频率之间的差异来校正预期邻近评分。其中一项二项式测试说明了数据集中顺式片段的总数,以及至少一次读取所覆盖的顺式片段总数。另一项二项式测试说明了数据集中反式片段的总数,以及至少一次读取所覆盖的反式片段总数。
[0272]
使用环化染色体构象捕获(4c)数据在感兴趣区域中检测染色体易位的实施例
[0273]
在该实施例中,选择感兴趣区域。感兴趣区域通常包含癌基因或抑癌基因,并且通常发现该区域在特定类型的癌症中被重排。接下来,使用设计用于侧接至少一个频繁易位位点的引物在感兴趣区域进行4c实验(krijger等人2019)。可选地,可以将独特分子标识符(umi)连接到引物上,以确保连接被独立捕获(schwartzman等人,2016)。在涉及连接产物pcr扩增的4c(类)实验中不使用umi的情况下,优选首先过滤片段的连接频率以去除pcr重复,这例如可以通过下游分析中的数据二值化来完成(即,仅区分捕获的(1)和未捕获的(0)片段)。因此,一旦产生的读取映射到参考基因组,就可以根据映射到每个片段的读取数来计算每个片段的连接频率。如果不使用umi,则至少一次读取所覆盖的片段连接频率设置为1,其余设置为0(即二值化,仅考虑独立连接的片段)。
[0274]
相邻片段的连接频率可以例如通过以每个片段为中心的高斯内核来聚集,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb和3mb或任何其他合适的值。这里kb表示千碱基,mb表示兆碱基。
[0275]
接下来,将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。换言之,将源自顺式染色体的第一片段的连接频率分配给源自顺式染色体的第二随机选择片段,并且将第二片段的连接频率分配给第一片段。通过该动作,第一片段和第二片段的原始连接频率分别改写成第二片段和第一片段的连接频率。
[0276]
类似地,将源自反式染色体的每个片段的连接频率与另一个来自反式染色体的随机选择片段交换。
[0277]
每个片段及其邻域交换的连接频率由以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。交换过程重复多次(通常n=1000),以形成数据集中每个片段的预期聚集连接评分集合。从该集合中,可以计算每个片段预期聚集连接评分的平均值和标准差。最后,将每个片段的观测聚集连接评分与相应片段的预期聚集连接评分的平均值和标准差进行比较,以计算每个片段的z-得分(或p值,如果优选)。z-得分(或p值)鉴定观测聚集连接评分显著增加的片段。
[0278]
在某些实施方式中,感兴趣区域中的结构变化检测实验例如可如下进行:
[0279]
1.选择需要进行结构完整性测试的感兴趣区域。
[0280]
2.使用设计用于侧接频繁易位位点的引物在感兴趣区域进行4c实验(krijger等人,2019)。
[0281]
3.可选地,将umi连接到引物上以识别独立连接的片段(schwartzman等人,2016)。
[0282]
4.将捕获读取映射到参考基因组。
[0283]
5.根据映射到每个片段的读取数计算每个片段的连接频率。
[0284]
6.如果未使用umi,则将至少一次读取所覆盖的片段的连接频率设置为1,其余片段设置为0(即二值化)。
[0285]
7.使用以每个片段为中心的高斯内核聚集相邻片段的连接频率,以形成观测聚集连接评分。例如,可以将邻域参数设置为200kb、750kb和3mb。然而,可以考虑任何期望的邻域参数。
[0286]
8.将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。
[0287]
9.将源自反式染色体的每个片段的连接频率与另一个来自反式染色体的随机选择片段交换。
[0288]
10.每个片段及其相邻片段交换的连接频率用以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0289]
11.多次重复交换过程(通常n=1000),以形成数据集中每个片段的聚集连接评分集合。
[0290]
12.可选地,将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。例如,该区域可以距离感兴趣区域+/-10mb。然而,可根据需要选择任何区域大小。该步骤可用于从分析中排除由于与感兴趣区域线性相邻而可能具有高显著性得分的观测聚集连接评分。
[0291]
13.使用数据集中每个片段聚集连接评分集合,计算数据集中每个片段的预期聚集连接评分的平均值和标准差。
[0292]
14.将每个片段的观测聚集连接评分与其预期聚集连接评分的平均值和标准差进
行比较,以计算z-得分(和/或p值,如果优选)。
[0293]
15.z评分高于某一阈值(例如7)的片段可被认为与感兴趣区域的基因组重排有关。类似地,p值低于某一阈值(例如0.1)的片段可被认为参与感兴趣区域的基因组重排。
[0294]
使用靶向位点扩增(tla)数据在感兴趣区域中检测染色体易位的实施例
[0295]
在该实施例中,选择感兴趣区域。感兴趣区域通常包含抑癌基因或抑肿瘤基因,并且通常发现该区域可在特定类型的癌症中被重排。接下来,使用设计用于侧接频繁易位位点或多个频繁易位位点的引物在感兴趣区域中进行tla实验(hottenot等人,2017)。一旦捕获的读取映射到参考基因组,就可以根据映射到每个片段的读取数来计算每个片段的连接频率。至少一次读取所覆盖的片段连接频率设置为1,其余设置为0(即二值化)
[0296]
相邻片段的连接频率可以通过以每个片段为中心的高斯内核来聚集,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb、3mb或任何其他合适的值。
[0297]
接下来,将源自顺式染色体的多个片段的聚集或未聚集连接频率与来自顺式染色体另一个随机选择选择片段交换。类似地,将源自反式染色体的多个片段的连接频率与另一个来自反式染色体的随机选择片段交换。每个片段及其相邻片段交换的连接频率由例如应用以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。交换过程重复多次(通常n=1000),以形成数据集中每个片段的可能聚集连接评分集合。从该集合中,可以计算预期聚集连接评分的平均值和标准差。最后,将每个片段的观测聚集连接评分与预期聚集连接评分的平均值和标准差进行比较,以计算每个片段的z-得分(或p值,如果优选)。z-得分(或p值)鉴定观测聚集连接评分显著增加的片段。
[0298]
在某些实施方式中,感兴趣区域中的结构变化检测实验例如可如下进行:
[0299]
1.选择需要进行结构完整性测试的感兴趣区域。
[0300]
2.使用设计用于侧接至少一个频繁易位位点的引物在感兴趣区域中进行tla实验(hottenot等人,2017)。
[0301]
3.将捕获读取映射到参考基因组。
[0302]
4.将至少一次读取所覆盖的片段的连接频率设置为1,其余片段设置为0(即二值化)。
[0303]
5.使用以每个片段为中心的高斯内核聚集相邻片段的连接频率,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb、3mb或任何其他合适的值。
[0304]
6.将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。
[0305]
7.将源自反式染色体的每个片段的连接频率与另一个来自反式染色体的随机选择片段交换。
[0306]
8.每个片段及其相邻片段交换的连接频率用以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0307]
9.多次重复交换程序(通常n=1000),以形成数据集中每个片段的预期聚集连接评分。
[0308]
10.计算数据集中每个片段的预期聚集连接评分的平均值和标准差。
[0309]
11.将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。该区域通常离感兴趣区域+/-10mb。这排除由于与感兴趣区域线性相邻而可能被评估的观测聚集连接
评分。
[0310]
12.将每个片段的观测聚集连接评分与其预期聚集连接评分的平均值和标准差进行比较,以计算z-得分(和p值,如果优选)。
[0311]
13.z-得分高于某一阈值(例如7)的片段可被认为与感兴趣区域的基因组重排有关。
[0312]
使用hi-c数据在感兴趣区域中检测染色体易位的实施例
[0313]
hi-c数据提供了细胞群体中染色质相互作用组的全基因组视图(lieberman-aiden等人,2009)。hi-c数据不描述代表感兴趣区域的选定片段(所谓的“视点”)与基因组中任何其他片段之间发生的3d相互作用(如4c或tla中所进行的,也称为一对全(one vs all)策略),而是表示基因组中每个片段与基因组中的任何其他片段之间的相互作用(也称为全对全(all vs all)策略)。因此,可以将hi-c数据分割成许多感兴趣区域,每个区域可以使用本文公开技术独立分析其结构完整性。为此,可以首先将获得的hi-c测序读取映射到参考基因组。接下来,可以选择发现连接到所选感兴趣区域的读取。接下来,使用所选择的读取,可以根据映射到每个片段的所选读取的数量来计算每个片段的连接频率。
[0314]
相邻片段的连接频率可以例如通过以每个片段为中心的高斯内核来聚集,以形成观测聚集连接评分。邻域参数(即长度规模)可以设置为200kb、750kb和3mb,但也可以考虑其他大小。
[0315]
接下来,将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。类似地,源自反式染色体的每个片段的连接频率可与另一个来自反式染色体的随机选择片段交换。每个片段及其相邻片段交换的连接频率由例如以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0316]
上述交换过程可重复多次(通常n=1000),以形成数据集中每个片段的聚集连接评分集合。从该集合中,可以计算每个片段预期聚集连接评分的平均值和标准差。最后,将每个片段的观测聚集连接评分与预期聚集连接评分的平均值和标准差进行比较,以计算每个片段的z-得分或p值。该得分识别了观测聚集连接评分显著增加的片段。
[0317]
在某些实施方式中,感兴趣区域中的结构变化检测实验例如可如下进行:
[0318]
1.对感兴趣的细胞/组织进行hi-c实验(lieberman aiden等人,2009)。
[0319]
2.将捕获读取映射到参考基因组。
[0320]
3.定义要进行结构完整性测试的感兴趣基因组区域。
[0321]
4.选择发现与感兴趣区域连接的读取。
[0322]
5.使用例如以每个片段为中心的高斯内核聚集相邻片段的连接频率,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb和3mb,但也可以考虑其他类似的大小。
[0323]
6.将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。
[0324]
7.将源自反式染色体的每个片段的连接频率与另一个来自反式染色体的随机选择片段交换。
[0325]
8.每个片段及其相邻片段交换的连接频率用例如以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0326]
9.多次重复交换程序(通常n=1000),以形成数据集中每个片段的预期聚集连接
评分。
[0327]
10.计算数据集中每个片段的预期聚集连接评分的平均值和标准差。
[0328]
11.将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。例如,这可用于距离感兴趣区域+/-10mb的基因组区域。该可选步骤可用于排除由于与感兴趣区域线性相邻而可能被评估的观测聚集连接评分。
[0329]
12.将每个片段的观测聚集连接评分与其预期聚集连接评分的平均值和标准差进行比较,以计算得分,例如z-得分(和/或p值,如果优选)。
[0330]
得分高于某一阈值(例如z-得分7)的片段可被认为与感兴趣区域的基因组重排有关。
[0331]
使用hi-c数据的全基因组染色体易位检测实施例
[0332]
hi-c数据提供了细胞群体中染色质相互作用组的全基因组视图(lieberman-aiden等人,2009)。hi-c数据不描述代表感兴趣区域的选定片段(所谓的“视点”)与基因组中任何其他片段之间发生的3d相互作用(如4c或tla中所进行的,也称为一对全(one vs all)策略),而是表示基因组中每个片段与基因组中的任何其他片段之间的相互作用(也称为全对全(all vs all)策略)。因此,通过修改所描述方法和一些细小改动,可以利用hi-c数据来提供整个基因组结构完整性的完整图片。为此,可以首先将获得的hi-c测序读取映射到参考基因组。接着,选择连接片段对。接下来,使用所选择的片段对,可以计算每个片段对的连接频率。这基本上形成了一个矩阵,该矩阵保存了观察基因组中每个dna片段对组合的一对彼此连接的dna片段的频率。
[0333]
相邻片段对的连接频率可以例如通过以每个片段对为中心的2d高斯内核来聚集,以形成观测聚集连接评分。邻域参数(即长度规模)可以设置为200kb、750kb和3mb,但也可以考虑其他大小。
[0334]
接下来,每个片段对的连接频率可以与另一随机选择的相关片段对交换(见图4)。每个片段对及其相邻片段交换的连接频率由例如以每个片段对为中心的高斯内核聚集,以计算每个片段对的随机化聚集连接评分。
[0335]
上述交换过程可重复多次(通常n=1000),以形成数据集中每个片段对的聚集连接评分集合。从该集合中,可以计算每个片段对预期聚集连接评分的平均值和标准差。最后,将每个片段对的观测聚集连接评分与预期聚集连接评分的平均值和标准差进行比较,以计算每个片段对的z-得分或p值。该得分识别了观测聚集连接评分显著增加的片段对。
[0336]
在某些实施方式中,结构变化检测实验可如下进行:
[0337]
1.对感兴趣的细胞/组织进行hi-c实验(lieberman aiden等人,2009)。
[0338]
2.将捕获读取映射到参考基因组。
[0339]
3.选择连接的片段对。
[0340]
4.使用例如以每个片段对为中心的高斯内核聚集相邻片段对的连接频率,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb和3mb,但也可以考虑其他类似的大小。
[0341]
5.将每个片段对的连接频率与另一个随机选择的相关dna片段对交换。
[0342]
6.每个片段对及其相邻片段对交换的连接频率用例如以每个片段对为中心的2d高斯内核聚集,以计算每个片段对的随机化聚集连接评分。
[0343]
7.多次重复交换程序(通常n=1000),以形成数据集中每个片段对的预期聚集连接评分。
[0344]
8.计算数据集中每个片段对的预期聚集连接评分的平均值和标准差。
[0345]
9.将驻留在感兴趣区域附近的片段对的观测聚集连接评分设置为零。例如,这可用于距离感兴趣区域+/-10mb的基因组区域。该可选步骤可用于排除由于与感兴趣区域线性相邻而可能被评估的观测聚集连接评分。
[0346]
10.将每个片段对的观测聚集连接评分与其预期聚集连接评分的平均值和标准差进行比较,以计算得分,例如z-得分(和/或p值,如果优选)。
[0347]
11.得分高于某一阈值(例如z-得分7)的片段对可被认为与感兴趣区域的基因组重排有关。
[0348]
使用捕获hi-c数据在感兴趣区域中检测染色体易位的实施例
[0349]
可以采用捕获hi-c实验(dryden等人,2014)或使用捕获探针的类似实验下拉并提取感兴趣基因组区域的序列(例如跨越整个基因座,或细分为多个部分的基因座),这些序列连接到细胞核中与感兴趣基因组区域序列邻近的片段,以帮助识别感兴趣基因组区域中可能的重排伴侣和断裂点。例如,涉及感兴趣基因组区域的相互易位将使该区域的一部分与一个衍生染色体融合,而感兴趣基因组区域的另一部分与另一个衍生染色体融合。结果,位于重排断裂点一侧的感兴趣基因组区域的部分将在断裂点处和朝向融合的反式染色体一侧显示显著增加的连接频率,而位于重排断裂点另一侧的感兴趣基因组区域的部分将显示从断裂点向融合的反式染色体另一侧显著增加的连接频率。通过使用本文公开技术选择性分析感兴趣基因组区域的不同部分的连接产物,可以估计或甚至确定两个重排基因座中的断裂点位置。
[0350]
一旦捕获的读取映射到参考基因组,就可以根据映射到每个片段的读取数来计算每个片段的连接频率。如果执行配对末端测序,则可根据感兴趣区域中连接的基因组部分(或片段)将测序读取拆分为多个数据集。
[0351]
相邻片段的连接频率可以例如通过以每个片段为中心的高斯内核来聚集,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb和3mb,但也可以考虑其他大小。
[0352]
接下来,将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。类似地,源自反式染色体的每个片段的连接频率可与另一个来自反式染色体的随机选择片段交换。每个片段及其相邻片段交换的连接频率由例如以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0353]
上述交换过程可重复多次(例如n=1000),以形成数据集中每个片段置换的聚集连接评分集合。从该集合中,可以计算预期聚集连接评分的平均值和标准差。
[0354]
最后,可将每个片段的观测聚集连接评分与预期聚集连接评分的平均值和标准差进行比较,以计算每个片段的得分,例如z-得分或p值。该分数可识别具有显著增加的观测聚集连接评分的片段。
[0355]
在某些实施方式中,感兴趣区域中的结构变化检测实验例如可如下进行:
[0356]
1.选择需要进行结构完整性测试的感兴趣区域。
[0357]
2.使用设计用于覆盖至少一个频繁易位基因组位点的一组引物在感兴趣区域中进行捕获hic实验(dryden等人,2014)。
[0358]
3.将捕获读取映射到参考基因组。
[0359]
4.可(在配对末端测序的情况下)根据与其连接的感兴趣基因组位点将映射的读取拆分为多个数据集。使用连接到所选感兴趣区域的片段数据集执行以下步骤。
[0360]
5.任选将至少一次读取所覆盖的片段的连接频率设置为1,其余片段设置为0(即二值化)。
[0361]
6.使用例如以每个片段为中心的高斯内核聚集相邻片段的连接频率,以形成观测聚集连接评分。邻域参数可以设置为200kb、750kb和3mb,但也可以考虑其他大小。
[0362]
7.将源自顺式染色体的每个片段的连接频率与另一个来自顺式染色体的随机选择片段交换。
[0363]
8.将源自反式染色体的每个片段的连接频率与另一个来自反式染色体的随机选择片段交换。
[0364]
9.每个片段及其相邻片段交换的连接频率用例如以每个片段为中心的高斯内核聚集,以计算每个片段的随机化聚集连接评分。
[0365]
10.多次重复交换过程(通常n=1000),以形成数据集中每个片段的置换聚集连接评分集合。
[0366]
11.计算来自置换聚集连接得分集合的数据集中每个片段的预期聚集连接评分的平均值和标准差。
[0367]
12.将驻留在感兴趣区域附近的片段的观测聚集连接评分设置为零。例如,该区域可以距离感兴趣区域+/-10mb。这排除由于与感兴趣区域线性相邻而可能被评估的观测聚集连接评分。
[0368]
13.将每个片段的观测聚集连接评分与其预期聚集连接评分的平均值和标准差进行比较,以计算得分,例如z-得分和/或p值(如果优选)。
[0369]
14.得分高于某一阈值(例如z-得分7)的片段可被认为与感兴趣区域的基因组重排有关。
[0370]
15.如果在步骤4中创建了多个数据集(使用不同感兴趣区域),则对至少一些具有适用于该数据集的感兴趣基因组区域的其他数据集重复步骤5-14。组合不同数据集的结果以获得关于重排位置的更详细信息。
[0371]
在本公开中,描述了一种处理来自邻近连接测定的数据以检测异常(例如染色体重排)的方法。用作该分析方法起点的数据可以是通过执行邻近连接测定、对该邻近连接测定的邻近连接片段进行测序以及将经测序的邻近连接片段映射到参考基因组而获得的数据集。
[0372]
因此,该分析的起点可以是包含多个映射到参考基因组的经测序的邻近连接片段的数据集。此外,可以根据手头的应用或者根据用户想要评估的任何猜想来选择感兴趣基因组区域。
[0373]
在某些实施方式中,考虑顺式dna片段的邻近评分与其离参考基因组中感兴趣区域的线性染色体距离之间的关系,以更严格地估计顺式染色体中dna片段的预期聚集连接评分,并搜索顺式染色体重排,例如缺失或倒位或插入,如下文进一步详述。为此,对于源自顺式染色体的每个dna片段,相关的dna片段基于它们与感兴趣区域的相似线性距离或基于非线性距离函数以概率定义,该非线性距离函数距离感兴趣区域dna片段越远越小(geeven
等人,2018)。在置换过程中,随机选择相关的dna片段,以估计顺式染色体中每个dna片段的预期聚集连接评分。
[0374]
在某些实施方式中,通过搜索来自顺式染色体上他处或来自具有高于某一阈值的邻近显著性得分的反式染色体的dna片段,检测源自顺式染色体或反式染色体上他处的dna序列基因组插入到感兴趣基因组区域(或邻近感兴趣基因区域的序列)中。
[0375]
在某些实施方式中,通过最初校正顺式染色体中dna片段的预期聚集邻近评分,然后搜索具有低于特定阈值(该阈值指示这些dna片段缺失)的负显著性得分的基因组dna片段,识别涉及感兴趣基因组区域(或感兴趣基因区域的邻近序列)的dna序列的基因组缺失。或者,通过搜索与感兴趣基因组区域相比显著性得分高于某一阈值(表示这些dna片段位于顺式染色体上缺失部分对侧)的基因组dna片段来识别基因组缺失,这是缺失紧靠感兴趣基因组区域的结果。
[0376]
类似地,通过最初校正顺式染色体中dna片段的预期聚集连接评分,然后在感兴趣基因组区域的顺式染色体中搜索基因组dna片段(所述基因组dna片段具有高于特定阈值的正显著性得分),以及感兴趣基因组区域的顺式染色体中的基因组dna片段(具有低于代表倒位基因组区域近端的特定阈值的负显著性得分),来识别涉及感兴趣区域部分和与感兴趣基因组区域邻近的序列的dna序列的基因组倒位。
[0377]
在某些实施方式中,为了独立确认检测到的结构变化,特定dna片段上结构变化的预估显著性得分可以帮助识别结构变化存在的额外证据,特别是通过促进在邻近(连接)数据集中发现以碱基对分辨率表示参考基因组中彼此不相邻的两个序列融合的读取。
[0378]
在某些实施方式中,可以通过根据源自感兴趣区域的连接dna片段内同时发生的单核苷酸改变来连接感兴趣区域中dna片段,检测单倍型特异性结构变化。使用这些连接,形成单倍型特异性邻近连接数据集。然后按照所公开的技术处理每个数据集,以识别单倍型特异性结构变化。
[0379]
在某些实施方式中,单倍型特异性结构变异可以通过分析包含评分为涉及结构变化的dna片段和来自发现其邻近的感兴趣基因组区域的dna片段的成对读取来检测,每个读取对用于等位基因分辨遗传变异,使得结构变异可以被单倍型抵消。
[0380]
本发明的一些或所有方面可以适合于以软件、特别是计算机程序产品的形式实现。计算机程序产品可以包括存储在非临时计算机可读介质上的计算机程序。此外,计算机程序可以由传输介质(例如光纤电缆或空气)携带的信号(例如光信号或电磁信号)表示。计算机程序可以部分或全部具有适合于由计算机系统执行的源代码、目标代码或伪代码的形式。例如,代码可以由一个或多个处理器执行。
[0381]
如本文所述,邻近测定,例如邻近连接测定,适合于识别重排和候选重排伴侣。本发明人已经认识到,用这种测定检测重排并不总是表明重排发生在感兴趣基因组区域内。如本领域技术人员将理解的,在感兴趣基因组区域之外的重排可能不会对感兴趣基因区域产生功能性后果。如本文进一步讨论的,本发明人认识到,包含感兴趣基因组区域的5'端侧接基因组片段和3'端侧接片段的基因组片段的邻近连接产物富集提高了识别涉及感兴趣基因组区域内断裂点的染色体重排的准确性。具体而言,富集策略可以设计为使固有噪声最小化,这反过来支持下游分析,以更好地区分感兴趣基因组区域内的真染色体重排(“真阳性调用(call)”)和感兴趣区域外的染色体重排(“假阳性调用”)。更重要的是,应该设计
富集策略,以使人们能够最好区分在感兴趣基因组区域内具有染色体断裂点和在顺式(在同一染色体上)但在感兴趣基因组区域外具有染色体断裂点的的染色体重排,从而区分相关和非相关事件。
[0382]
染色体重排的假阳性调用可能由于各种原因而发生,其中一个原因是偶尔不希望的探针或引物杂交到基因组其他地方的脱靶序列。结果,脱靶邻近连接产物将被富集、测序和定位,因此可以显示携带脱靶杂交序列的染色体片段上邻近连接产物的聚集。这种信号的聚集可能被错误地识别为具有染色体重排(假阳性调用)。
[0383]
已经制定了多种策略来解决这种不期望的影响。一种策略是使用预期不会携带涉及感兴趣染色体区域重排的对照个体。鉴定对照样品中相同染色体重排是识别这些调用为假阳性的充分证据。在这种情况下,覆盖重排的相应染色体片段可以被列入黑名单。另一种防止由脱靶探针或引物杂交以及随后脱靶染色体邻近产物富集引起的重排的假阳性调用的策略是识别引起脱靶杂交的单个探针或引物,并将其从靶向感兴趣染色体区域的探针或引物组中物理或计算机扣除。
[0384]
假阳性调用的另一个来源来自研究样本基因组中存在的拷贝数变异。尽管潜在的生物学原因不同于脱靶探针或引物杂交,但基因组中拷贝数变异增加的基因组片段可能显示出邻近连接产物的聚集。再次,这种信号的聚集可能被错误地识别为相关染色体重排(假阳性调用)。为了解决这一问题,可以分析来自同一样本上定义的其他感兴趣区域的邻近联接数据集。为此,可以通过查询是否从同一样本中的不同感兴趣区域识别出相同的染色体重排来识别拷贝数变化的存在,但这并不总是足够的。
[0385]
如上所述,邻近测定可以容易地检测染色体重排。然而,本文所述的例子表明,这种测定并不总是能区分在感兴趣基因组区域内具有断裂点连接(相关)和在感兴趣基因组区域外具有染色体断裂点连接(不相关)的事件。令人惊讶的是,在许多染色体断裂点位于感兴趣的基因组区域之外的情况下,发现了明显高于预期的聚集在融合基因组伴侣上的核邻近产物,导致检测到该事件并调用为“阳性”。这些例子进一步证明,当断裂点与感兴趣区域顺式(同一染色体上)相距数兆碱基时,这种假阳性调用甚至也会发生。对于许多应用程序,区分这两种场景至关重要。
[0386]
本领域技术人员已知大量基因,其在发生突变时(例如由于重排)与疾病如癌症相关。为了让医生准确地诊断或预测所述疾病,重要的是了解重排发生在与感兴趣基因组区域相关的位置。例如,当搜索产生致癌融合基因产物的融合基因时,优选将染色体断裂点映射到基因内部的位置。作为另一个例子,当搜索可能将原癌基因置于将其表达水平改变为致癌活性水平的新转录调控dna序列影响下的染色体重排时,优选将染色体重排断裂点映射到足够接近原癌基因的染色体位置,以预期其转录调节的改变。
[0387]
发明人已经认识到,可以改进现有技术的方法,以使得真正“阳性”调用的可靠性增加。因此,本公开的一个方面提供了用于确认样品(特别是患者样品,例如肿瘤细胞样品)是否包括临床相关染色体重排的方法。本公开进一步提供了用于鉴定指示特定疾病、预后或预测治疗反应的染色体重排的方法。
[0388]
本公开提供了用于确认染色体断裂点连接存在的方法,所述染色体断裂点连接将候选重排伴侣融合到感兴趣基因组区域内的位置。如本文所用,确认染色体断裂点连接的存在还指检测将候选重排伴侣融合到感兴趣的基因组区域内的位置的染色体断裂点连接
的存在。优选地,所述方法包括确定参考基因组中的感兴趣基因组区域。在一些实施方式中,感兴趣基因组区域在100bp至1mb之间,例如从1kb至10,00kb。
[0389]
在优选实施方式中,感兴趣基因组区域是指编码基因开放阅读框的dna序列。本领域技术人员将容易理解,位于开放阅读框内的断裂点融合可能影响所述基因的功能。取决于重排的性质,重排可导致例如由感兴趣基因组区域编码的蛋白质、包含由感兴趣基因组区域编码的蛋白质的一部分和由重排伴侣编码的蛋白质部分的融合蛋白的过早截断,以及包含由感兴趣基因组区域编码的蛋白质的至少一部分以及来自现在编码“新(neo)
”‑
蛋白序列的重排伴侣的框外序列的新颖蛋白质。
[0390]
在一个优选实施方式中,感兴趣基因组区域指基因。本领域技术人员将容易理解,位于基因序列内的断裂点融合可能影响所述基因的功能。除了上述关于在开放阅读框中发生的重排影响之外,重排还可以影响例如mrna的表达和/或转录。例如,染色体重排可能会使基因受到新的转录调控dna序列的影响,从而改变基因的表达水平。具有转录调节潜力的跨越基因组区间的序列对于每个基因大小会有所不同。考虑如染色体构象研究检测的,包含优选在感兴趣组织或细胞类型中的靶基因结构域或拓扑关联域(tad),可以提高检测相关染色体重排的分析效率。结构域或tad是染色体片段,其中序列优先彼此接触,它们两侧有边界,阻止基因与结构域外的转录调控序列接触并受其调控。因此,位于结构域外的染色体断裂点不太可能影响靶基因的表达。如果结构域或tad未定义,可以确定感兴趣基因组区域,例如,作为靶基因启动子上游的一兆碱基和下游的一兆碱基,因为很少有转录调控序列可以在比一兆碱基更远的距离上起作用。本领域技术人员还意识到,当在基因沙漠的情况下(即,没有或很少有基因围绕靶基因的基因组间隔),转录调控序列可能离基因更远。基因沙漠通常包含转录调控序列,这些序列可以长距离作用于线性分离的基因。
[0391]
优选地,感兴趣基因组区域是本领域技术人员已知发生重排的基因或开放阅读框的子序列。例如,感兴趣基因组区域优选地指断裂点簇区域。这些簇是本领域熟知的。特别地,本领域技术人员知道与特定病症相关的潜在断裂点簇。在一些实施方式中,所述方法适于确定是否在与特定病症相关的断裂点簇内发生重排。断裂点簇区域的一个例子是人类18号染色体上编码bcl2基因3'utr区域中长175bp的最3'的外显子,占bcl2基因所有断裂的50%(tsai&lieber,bmcgenomics(2010)11:1)。断裂点簇区域的另一个例子是人类11号染色体上mll基因的外显子9和外显子13之间长7466bp的染色体区域(burmeister等,leukemia(2006)20445-457)。
[0392]
该方法包括执行邻近测定以生成多个邻近联接产物。在一些实施方式中,该测定是产生多个邻近连接分子的邻近连接测定(参见例如图1)。这种邻近连接测定在本文中进一步描述。在示范性邻近连接测定中,用限制性酶消化交联的dna(例如甲醛交联的),并在有利于交联dna片段之间邻近连接的条件下重新连接,以产生邻近连接的分子。优选在连接后逆转交联。
[0393]
在一些实施方式中,邻近连接测定包括:
[0394]
a)提供交联dna的样品;
[0395]
b)片段化交联的dna;
[0396]
c)连接片段化的交联dna以获得邻近连接的分子;
[0397]
d)逆转交联;
[0398]
e)任选地将步骤d)的dna片段化(例如用限制性酶或超声处理)。在一些实施方式中,所述方法还包括:
[0399]
f)将步骤d)或e)的片段化dna与至少一个衔接子连接,和
[0400]
g)使用与靶核苷酸序列杂交的至少一种引物扩增步骤d)或e)的包含靶核苷酸序列的连接的dna片段,或使用与所述靶dna序列杂交的至少一种引物和与所述至少一个衔接子杂交的至少一种引物扩增步骤f)的连接的dna片段。
[0401]
优选地,该方法包括提供交联dna样品用于邻近测定。
[0402]
在一些实施方式中,该方法包括富集邻近联接产物,其包括基因组片段,该基因组片段包含感兴趣基因组区域或侧接感兴趣基因区域的序列。本领域技术人员知道多种靶向dna富集策略。通常,这种方法依赖于寡核苷酸(如探针或引物)与感兴趣序列的杂交。
[0403]
在一个实施方式中,该方法包括富集包含侧接感兴趣基因组区域5’端的序列的基因组片段的邻近连接产物,以及富集包含侧接感兴趣基因组区域3’端的序列的基因组片段的邻近连接产物。可对邻近连接的产物进行测序,以产生与所述包含侧接感兴趣基因组区域的5’或3’端的序列的基因组片段邻近的基因组片段序列的读数,可映射到参考序列。“侧接序列”是指与感兴趣区域相邻的序列。侧接序列可以直接或间接邻近感兴趣区域。
[0404]
在一个实施方式中,该方法包括提供至少一种寡核苷酸探针或引物,其至少部分与侧接感兴趣基因组区域5'区的序列互补,和/或提供至少一种寡聚核苷酸探针或引物,其至少部分与侧接感兴趣基因组区域3'区的序列互补。在一些实施方式中,探针和引物与独特靶序列互补,以防止与重复dna杂交。寡核苷酸探针可以附着在固体表面上,或包含允许在固体表面(如链霉亲和素珠)上捕获的标签(如生物素)。在一些实施方式中,衔接子序列可以连接到片段化dna。然后,可用一个互补于侧接感兴趣基因组区域的序列的引物和另一个互补于衔接子序列的引物pcr扩增。或者,衔接子序列可用于生成测序读取。探针和引物设计是本领域技术人员熟知的。优选地,寡核苷酸探针和引物与感兴趣基因组区域上游或下游1bp至1mbp之间的序列互补。或者,侧接可能指的是基因组区域或序列,其距离问题染色体为其长度的0.5%或更小。在一些实施方式中,可以使用侧接感兴趣基因组区域的探针/引物组。
[0405]
该方法还包括基于所述基因组片段与感兴趣基因组区域或与侧接感兴趣基因组区域的序列的邻近频率,识别至少一个基因组片段作为候选重排伴侣。如本文进一步所述,所述方法可包括富集包含i)至少部分感兴趣基因组区域和ii)与感兴趣基因组区域邻近的基因组片段的邻近连接产物。优选地,该方法富集感兴趣基因组区域的至少一部分。虽然通过富集包含侧接感兴趣基因组区域的序列的邻近连接分子来确认感兴趣基因组区内断裂点连接的存在,候选重排伴侣的鉴定可以基于包括感兴趣基因组区域或侧接感兴趣基因区域的序列的测序读取进行。
[0406]
在示例性实施方式中,通过使用互补寡核苷酸探针来下拉和富集涉及感兴趣基因组区域的核邻近产物,邻近测定可针对感兴趣的特定基因组区域。或者,通过使用互补寡核苷酸引物(引物)对涉及感兴趣基因组区域的染色体邻近产物进行线性或指数扩增和富集,染色体邻近测定可针对感兴趣的特定基因组区域。富集后,对邻近产物进行测序,并将序列读取映射到参考基因组。染色体重排是基于基因组中他处基因组片段的鉴定而发现的,显示涉及感兴趣基因组区域的核邻近产物聚集明显高于预期。
[0407]
用于基于邻近频率识别候选重排伴侣的合适方法是本领域已知的,并在本文中描述。例如,可以使用感兴趣基因组区域接触情况的目视检查(参见,例如,simonis等人,2009;de vree等人,2014;和wo2008084405)。例如参见harewood等人,了解基于选择前1%高度相互作用的染色体内区域的方法(genome biology 201718:125)。另请参见本文描述的d
í
az等人2018和dixon等人2018中描述的方法。其他方法包括salsa、gothic、hiccompare、hifi、v4c、lachesis、hint、bin3c。mifsud描述了一种模型(gothic),用于从邻近连接数据中识别真实相互作用,还回顾了用于识别重排伴侣的其他众所周知的模型(plos one 2017 12(4):e0174744)。
[0408]
识别候选重排伴侣的优选方法如图1-6所示,本文称为plier。在一些实施方式中,识别一个或多个候选重排伴侣的方法包括:
[0409]
选择多个经测序的邻近连接的dna分子,所述dna分子包括映射到感兴趣基因组区域的序列;
[0410]
向基因组的多个基因组片段中的每一个分配(101)观测邻近评分,每个基因组片段的观测邻近评分指示在数据集中存在邻近感兴趣基因组区域且包括对应于基因组片段的序列的至少一个测序读取;
[0411]
基于多个基因组片段的观测邻近评分,将预期邻近评分分配(102)给所述多个基因组片段中至少一个基因组片段中的每一个,其中所述预期邻近评分包括所述多个基因组片段中至少一个邻近评分的预期值;和
[0412]
基于多个基因组片段中的所述至少一个基因组片段的观测邻近评分和多个基因组片段中的所述至少一个基因组片段的预期邻近评分,生成(103)多个基因组片段中的所述至少一个基因组片段参与染色体重排的可能性指示,和将所述基因组片段鉴定为候选重排伴侣。本文进一步描述了该方法的优选实施方式,图6提供了该方法特别优选实施方式。
[0413]
一旦识别了候选重排伴侣,该方法包括确定与所述包含侧接感兴趣基因组区域5’端的序列的基因组片段邻近的候选重排伴侣的基因组片段以及与所述包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的候选重排伴侣的基因组片段是重叠的还是线性分离的。
[0414]
邻近感兴趣基因组区域的第一部分或感兴趣区域的侧接区域的基因组片段将显示与邻近感兴趣基因组区域的第二部分或感兴趣区域的侧接区域的基因组片段的“混合”或“分裂”聚集。显示混合聚集的片段在本文中称为“重叠”,显示分裂聚集的片段称为“线性分离”。优选所述方法包括确定邻近感兴趣基因组区域的第一部分或感兴趣区域的侧接区域的候选重排伴侣的基因组片段和邻近感兴趣基因组区域的第二部分或感兴趣区域的侧接区域的候选重排伴侣的基因组片段,当映射到候选重排伴侣的参考序列时,是重叠的还是线性分离的。
[0415]
例如,可以分析源自侧接感兴趣基因组区域的上游和下游序列的邻近产物,以确定重排伴侣的分布。如果侧接基因组序列在重排伴侣的线性参考模板上显示联接产物的重叠(混合)聚集,这表明断裂点不在感兴趣基因组区域内。如果侧接基因组序列在重排伴侣的线性参考模板上显示分裂的聚集(本文也称为“转变”或“线性分离”),这表明断裂点位于感兴趣基因组区域内。关于重排伴侣,染色体断裂点位于基因组区段处,该区段标志着从源自侧接感兴趣基因组区域的上游序列的邻近产物到源自侧接感兴趣基因区域的下游序列
的邻近产品的聚集转变。如果仅一个侧接区域(即仅5’侧接序列或仅3’侧接序列)对重排伴侣贡献邻近产物,这表明不平衡的染色体重排或复杂染色体重排,其在感兴趣基因组区域内具有断裂点,并且其他侧接序列缺失或其与基因组中的另一个伴侣融合(参见例如,图9),以及外来dna的插入。
[0416]
在优选实施方式中,将与包含侧接感兴趣基因组区域3’端的序列的基因组片段邻近的基因组片段(例如,对应于候选重排伴侣)的序列位置和与包含侧接感兴趣基因组5’端的序列的基因组片段邻近的基因组片段(如对应于候选重排伴侣)的序列位置进行比较。所述候选重排伴侣基因组片段的线性分离指示所述感兴趣基因组区域内的染色体断裂点连接。在一些实施方式中,该方法包括分析在重排伴侣和侧接感兴趣基因的靶标5’和3’序列之间形成的富集邻近联接产物分别是否在含有重排伴侣的线性染色体模板上分离。这种线性分离是感兴趣基因内染色体断裂的证据。
[0417]
目测观察重叠和线性分离的一种方法是从与基因组片段相对应的序列读取中生成矩阵,其中一条轴表示对应于感兴趣基因组区域或侧接感兴趣基因组区域的序列的序列位置,另一条轴代表与感兴趣基因组区域或侧接感兴趣基因组区域的序列(例如候选重排伴侣)联接的基因组片段的序列位置。所述联接邻近产物可以叠加在所述矩阵上,使得所述矩阵内每个元素表示联接产物被发现包括感兴趣区域内或侧接的对应基因组区段以及与感兴趣区域内或侧接的所述对应基因组区段联接的基因组区段的次数。见例如图9b描述了位置4的重排。候选重排伴侣的序列在感兴趣基因组区域的位置“a”和“b”重叠。如本领域技术人员所清楚的,重叠的候选重排伴侣序列不要求包含“a”的邻近连接分子和包含“b”的邻近链接分子也必须包含相同或物理上重叠的重排伴侣序列。相反,本领域技术人员理解存在这样的序列混合。将这与下文所述的线性分离比较。
[0418]
如上所述,目测线性分离的一种方法是生成矩阵。如果轴上的一个或多个代表感兴趣的基因组区域和/或侧接感兴趣基因组区域的区域的序列位置的坐标显示来自重排伴侣的基因组区段邻近频率中的转变,则表示线性分离。特别是比较来自与感兴趣基因组区域和/或侧接感兴趣基因组区域的区域的基因组片段邻近的候选重排伴侣的基因组区段邻近频率,所述基因组区段使用本文公开的邻近测定富集。
[0419]
在一些实施方式中,还富集了包含感兴趣基因组区域的邻近联接产物。优选使用探针/引物来覆盖感兴趣基因组区域的重要部分,从而可以获得感兴趣基因区域的重要部分的邻近数据。如果根据相邻象限之间频率的最大差异和象限内频率的最小差异,矩阵可以在特定位置分为四个象限,则表示线性分离,这表示染色体断裂点。例如,参见图9b,其描述了位置1、2和3处的重排以及图9c中的例子。这些例子描述了一种可能的相互重排。
[0420]
当基因组片段(例如对应于候选重排伴侣)靠近例如侧接感兴趣基因组区域5’区的序列而不靠近侧接感兴趣基因组区域3’的序列时,也存在线性分离(反之亦然)。这种形式的线性分离可以通过识别轴上的一个或多个坐标在矩阵中目测,所述轴代表感兴趣基因组区域和/或侧接感兴趣基因组区域的区域的序列位置,其显示来自候选重排伴侣的基因组片段邻近频率的转变。在非相互重排的情况下,该转变是从候选重排伴侣基因组片段的特定邻近频率到候选重排伴侣序列的(统计显著)缺失。在一个示范性实施方式中,通过在单个象限中存在基因组片段(例如,对应于候选重排伴侣)以及在其他三个象限中(统计上显著)不存在候选重排伴侣序列,可以在蝴蝶图矩阵中目测这种形式的线性分离。见例如图
9d所述例子。
[0421]
在一些实施方式中,该方法包括将得分分配给邻近联接产物的混合(即,重叠)程度。在一些实施方式中,所分配的得分指示重排是相互或非相互染色体重排。
[0422]
如实施例所示,富集包括含侧接感兴趣基因组区域5’端的序列的基因组片段的邻近连接产物,以及包括含侧接感兴趣基因组区域3’端的序列的基因组片段的邻近连接产物,令人惊讶地能够确认导致在感兴趣基因组区域内断裂点连接的重排并减少“假阳性”(见图9a)。
[0423]
如上所述,方法还可包括富集包含i)至少部分感兴趣基因组区域和ii)与感兴趣基因组区域邻近的基因组片段的邻近连接产物。在一些实施方式中,该方法包括提供与感兴趣基因组区域至少部分互补的多个探针或引物。所述多个寡核苷酸探针/引物中的每一个可被导向感兴趣基因组区域的不同或重叠的子序列。在一些实施方式中,探针/引物组被设计成以每100kb、每10kb或每1kb至少一个探针/引物的间隔靶向基因组区域。此类方法可用于确定染色体断裂点连接的位置,将候选重排伴侣融合到感兴趣基因组区域内的位置,或者更确切地说,用于“精细映射”断裂点连接。
[0424]
在这样的实施方式中,所述方法还包括对所述邻近连接的dna分子进行测序,所述dna分子包括i)至少部分感兴趣基因组区域和ii)与感兴趣基因组区域邻近的基因组片段,以产生感兴趣测序读取的基因组区域。
[0425]
所述方法还可以包括映射染色体断裂点,其中所述映射包括检测包含至少部分感兴趣基因组区域并且具有重排伴侣序列线性分离的邻近连接的dna分子。如本领域技术人员所清楚的,所述方法可包括鉴定包含在线性序列中彼此最接近且具有重排伴侣序列的线性分离的基因组感兴趣区域片段的邻近连接分子。这可通过例如根据它们在感兴趣基因组区域的线性模板上的原始位置来组织邻近连接产物(包括感兴趣基因区域的至少一部分和与感兴趣基因组区域邻近的基因组片段,例如候选重排伴侣),通过例如滑动窗口法,研究感兴趣基因组区域上的线性组织如何与映射到重排伴侣的其邻近连接产物的线性位置相关。滑过感兴趣基因组区域后的位置标志着从在重排伴侣的线性模板上混合(即重叠)的邻近连接产物转变为在重排伴侣线性模板上分离的邻近连接产物的位置,划定了感兴趣基因组区域内的染色体断裂点位置。
[0426]
在一些实施方式中,染色体断裂点的映射包括为测序读取的至少一个子集生成矩阵,其中所述矩阵的一条轴代表感兴趣基因组区域和/或侧接感兴趣基因区域的序列的序列位置,而另一条轴代表候选重排伴侣的序列位置,其中所述矩阵是通过在所述矩阵上叠加所述测序读取而产生的,使得所述矩阵内的每个元素代表包含感兴趣基因组区域的基因组片段和来自重排伴侣的基因组片段的邻近联接的dna分子的频率。优选矩阵是蝴蝶图。参见图9中bcl2和myc基因中断裂点连接的映射。
[0427]
在一些实施方式中,该方法包括确定跨越断裂点的基因组区域序列,所述方法包括鉴定邻近连接的dna分子,所述dna分子包括i)感兴趣基因组区域断裂点邻近序列和ii)重排伴侣序列。本文所述方法的一个优点涉及从测序数据中存在的“噪声”读取中过滤“真实”融合读取的能力。标准的下一代测序方法允许主要基于频率差异(真实和噪声之间)和/或融合伴侣的现有知识进行过滤步骤。在本公开的一些方面,通过首先应用定位候选重排伴侣的plier算法,可以将“真实”融合读取与噪声分离。或者,除了plier算法之外,使用多
topologies.nat.genet.50,1151
–
1160.
[0437]
brant l,georgomanolis t,nikolic m,brackley ca,kolovos p,van ijcken w,grosveld fg,marenduzzo d,papantonis a.exploiting native forces to capture chromosome conformation in mammalian cell nuclei.mol syst biol.2016dec 9;12(12):891.
[0438]
cairns,j.,freire-pritchett,p.,wingett,s.w.,v
á
rnai,c.,dimond,a.,plagnol,v.,zerbino,d.,schoenfelder,s.,javierre,b.m.,osborne,c.,等人,(2016).chicago:robust detection of dna looping interactions in capture hi-c data.genome biol.jun 15;17(1):127
[0439]
chesi,a.,wagley,y.,johnson,m.e.,manduchi,e.,su,c.,lu,s.,leonard,m.e.,hodge,k.m.,pippin,j.a.,hankenson,k.d.,等人,(2019).genome-scale capture c promoter interactions implicate effector genes at gwas loci for bone mineral density.nat.commun.10,1260.
[0440]
choy,m.k.,javierre,b.m.,williams,s.g.,baross,s.l.,liu,y.,wingett,s.w.,akbarov,a.,wallace,c.,freire-pritchett,p.,rugg-gunn,p.j.,等人,(2018b).promoter interactome of human embryonic stem cell-derived cardiomyocytes connects gwas regions to cardiac gene networks.nat.commun.jun 28;9(1):2526.
[0441]
dao,l.t.m.,galindo-albarr
á
n,a.o.,castro-mondragon,j.a.,andrieu-soler,c.,medina-rivera,a.,souaid,c.,charbonnier,g.,griffon,a.,vanhille,l.,stephen,t.,等人,(2017).genome-wide characterization of mammalian promoters with distal enhancer functions.nat.genet.49,1073
–
1081.
[0442]
dekker,j.,rippe,k.,dekker,m.,and kleckner,n.(2002)capturingchromosome conformation.science.295,1306-1311
[0443]
denker a,de laat w.(2016).the second decade of 3c technologies:detailed insights into nuclear organization.genes dev.30:1357-82.
[0444]
de vree pjp,de wit e,yilmaz m,van de heijning m,klous p,verstegen mjam,wan y,teunissen h,krijger phl,geeven g,eijk pp,sie d,ylstra b,hulsman lom,van dooren mf,van zutven ljcm,van den ouweland a,verbeek s,van dijk kw,cornelissen m,das at,berkhout b,sikkema-raddatz b,van den berg e,van der vlies p,weening d,den dunnen jt,matusiak m,lamkanfi m,ligtenberg mjl,ter brugge p,jonkers j,foekens ja,martens jw,van der luijt r,ploos van amstel hk,van min m,splinter e,de laat w(2014).targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping.nature biotechnology.oct;32(10):1019-25.
[0445]
dryden,n.h.,broome,l.r.,dudbridge,f.,johnson,n.,orr,n.,schoenfelder,s.,...&assiotis,i.(2014).unbiased analysis of potential targets of breast cancer susceptibility loci by capture hi-c.genome research,24(11),1854-1868.
[0446]
homminga,i.,pieters,r.,langerak,a.w.,de rooi,j.j.,stubbs,a.,verstegen,m.,vuerhard,m.,buijs-gladdines,j.,kooi,c.,klous,p.,van vlierberghe,
p.,ferrando,a.a.,cayuela,j.m.,verhaaf,b.,beverloo,h.b.,horstmann,m.,de haas,v.,wiekmeijer,a.s.,pike-overzet,k.,staal,f.j.,de laat,w.,soulier,j.,sigaux,f.,and meijerink,j.p.(2011)integrated transcript and genome analyses reveal nkx2-1 and mef2c as potential oncogenes in t cell acute lymphoblastic leukemia.cancer cell.19,484-497.
[0447]
hottentot qp,van min m,splinter e,white sj.targeted locus amplification and next-generation sequencing.methods mol biol.2017;1492:185-196.
[0448]
hughes,j.r.,roberts,n.,mcgowan,s.,hay,d.,giannoulatou,e.,lynch,m.,de gobbi,m.,taylor,s.,gibbons,r.,and higgs,d.r.(2014).analysis of hundreds of cis-regulatory landscapes at high resolution in a single,high-throughput experiment.nat.genet.46,205
–
212.
[0449]
r.,migliorini,g.,henrion,m.,kandaswamy,r.,speedy,h.e.,heindl,a.,whiffin,n.,carnicer,m.j.,broome,l.,dryden,n.,等人,(2015).capture hi-c identifies the chromatin interactome of colorectal cancer risk loci.nat.commun.feb 19;6:6178
[0450]
javierre,b.m.,sewitz,s.,cairns,j.,wingett,s.w.,v
á
rnai,c.,thiecke,m.j.,freire-pritchett,p.,spivakov,m.,fraser,p.,burren,o.s.,等人,(2016).lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters.cell.nov 17;167(5):1369-1384
[0451]
kwak,e.l.,bang,y.j.,camidge,d.r.,shaw,a.t.,solomon,b.,maki,r.g.,ou,s.h.,dezube,b.j.,janne,p.a.,costa,d.b.,varella-garcia,m.,kim,w.h.,lynch,t.j.,fidias,p.,stubbs,h.,engelman,j.a.,sequist,l.v.,tan,w.,gandhi,l.,minokenudson,m.,wei,g.c.,shreeve,s.m.,ratain,m.j.,settleman,j.,christensen,j.g.,haber,d.a.,wilner,k.,salgia,r.,shapiro,g.i.,clark,j.w.,and iafrate,a.j.(2010)anaplastic lymphoma kinase inhibition in non-small-cell lung cancer.the new england journal of medicine.363,1693-1703.
[0452]
li,g.,ruan,x.,auerbach,r.k.,sandhu,k.s.,zheng,m.,wang,p.,poh,h.m.,goh,y.,lim,j.,zhang,j.,等人,(2012).extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation.cell 148,84
–
98.
[0453]
lieberman-aiden,e.,van berkum,n.l.,williams,l.,imakaev,m.,ragoczy,t.,telling,a.,amit,i.,lajoie,b.r.,sabo,p.j.,dorschner,m.o.,等人,(2009).comprehensive mapping of long-range interactions reveals folding principles of the human genome.science 326,289
–
293.
[0454]
martin,p.,mcgovern,a.,orozco,g.,duffus,k.,yarwood,a.,schoenfelder,s.,cooper,n.j.,barton,a.,wallace,c.,fraser,p.,等人,(2015).capture hi-c reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci.nat.commun.nov 30;6:10069.
smallcell lung cancer.the new england journal of medicine.370,2537-2539.
[0466]
shaw,a.t.,ou,s.h.,bang,y.j.,camidge,d.r.,solomon,b.j.,salgia,r.,riely,g.j.,varella-garcia,m.,shapiro,g.i.,costa,d.b.,doebele,r.c.,le,l.p.,zheng,z.,tan,w.,stephenson,p.,shreeve,s.m.,tye,l.m.,christensen,j.g.,wilner,k.d.,clark,j.w.,and iafrate,a.j.(2014)crizotinib in ros1-rearranged non-small-cell lung cancer.the new england journal of medicine.371,1963-1971.
[0467]
simonis,m.,klous,p.,splinter,e.,moshkin,y.,willemsen,r.,de wit,e.,van steensel,b.,and de laat,w.(2006).nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip(4c).nat.genet.38,1348
–
1354.
[0468]
van de werken h,landan g,holwerda s,hoichman m,klous p,chachik r,splinter e,valdes quezada c,y,bouwman b,verstegen m,de wit e,tanay a,de laat w.(2012).robust4c-seq data analysis to screen for regulatory dna interactions.nature methods,9:969-72.
[0469]
本文描述的实施例和实施方式用于说明而不是限制本发明。本领域技术人员将能够在不脱离本公开的精神和范围的情况下设计替代实施方式,如所附权利要求及其等价形式限定的。权利要求书中括号内的参考符号不得解释为限制权利要求书的范围。在权利要求或说明书中被描述为独立实体的项目可以结合所描述项目的特征的单个硬件或软件项目实施。
[0470]
实施例:
[0471]
基因组中的结构变异(sv)是癌症复发的标志。易位(染色体之间的基因组重排)尤其被发现是许多类型血液淋巴恶性肿瘤的复发驱动因素。在各种类型的实体肿瘤中,如肺癌、前列腺癌和软组织肉瘤中,它们也越来越受到重视,作为诊断、预后甚至预测参数来指导治疗选择。因此,在这些恶性肿瘤的常规诊断工作流程中越来越多地实施特定靶基因组的易位分析。诊断病理实践高度依赖于福尔马林固定和石蜡包埋(ffpe)程序。所得到的ffpe标本块提供了一种长期保存方法,特别适用于形态学评估,包括免疫组织化学和原位杂交技术(ish)。目前,荧光原位杂交(fish)是淋巴瘤ffpe样本易位检测的“金标准”。尽管这种方法在世界范围内普遍应用,并在许多情况下取得了成功,但它有各种局限性。fish评估依赖于足够的形态学。因此,经常损害形态学的粉碎性伪影(artifact)、固定不良、广泛坏死和细胞凋亡,通常无法进行可靠的解释。此外,尽管fish测定可以与免疫组织化学相同的自动化方式常规进行,但结果分析和重排检测主要是手动进行的,其需要大量劳动,容易出错且昂贵。此外,如果出现导致荧光信号复杂模式的罕见断裂点、多聚核糖体或缺失,fish评估可能是困难的、模棱两可或主观的
1,2
。常规使用的分离-fish方法无法识别易位伴侣,而融合-fish仅适用于已知易位伴侣的特定情况,如myc-igh易位。了解重排的确切组成经常是描述肿瘤进展行为及其亚分类的必要信息3。最后,fish分析不能多重化。
[0472]
最近,下一代测序(ngs)dna捕获方法已被引入ffpe样本中选定基因组中的重排检测,这使得可以检测碱基对分辨率的断裂点并识别易位伴侣基因
4-7
。然而,这种方法依赖于捕获明确的融合读取,当非独特序列侧接断裂点时,这可能是一个挑战8。这是一种常见的情况,尤其是恶性淋巴瘤中的易位,通常涉及免疫球蛋白和t细胞受体基因作为癌基因的易
位伴侣9。基于rna的检测方法是ffpe材料中重排检测的另一种方法,目前在日常实践中引入了用于那些导致嵌合或改变rna产物重排的方法,如软组织肿瘤的典型方法
10-12
。rna不如dna稳定,这有时会影响ffpe样本中基于rna的诊断方法性能
13
。此外,基于rna的检测方法无法检测非编码序列中的重排,这些序列通过调节置换效应驱动癌症。这在恶性淋巴瘤中最常见,其中免疫球蛋白和t细胞受体增强子序列介导还步未改变的癌基因的过度表达。总之,在日常诊断病理实践中,仍然需要更可靠地检测和准确表征ffpe样本中易位的方法。
[0473]
重要的是,福尔马林固定和病理组织处理中的(非计划的)dna片段化是邻近连接(或“染色体构象捕获”)方法中的强制性步骤。邻近连接方法最初是为了研究染色体折叠
14
而发明的,使用甲醛介导的固定,然后原位dna片段化和连接,以融合细胞核内最接近的dna片段。然后ngs和连接产物的定量分析可以提供细胞群体中序列对之间的接触频率的相对估计,从而能够分析复现性染色体折叠模式。决定一对dna序列之间的接触频率的最主要因素是它们在同一染色体上线性邻近,因此这种接触频率随着两条dna序列之间线性分离的增加呈指数衰减。有趣的是,基因组重排改变了染色体的线性序列,从而改变了邻近连接方法中产生的dna接触模式。基于这种理解,已经引入了邻近连接方法的变体作为鉴定基因组重排的强大技术
15-20
。最近在一项非盲研究中提供了邻近连接方法也可以检测ffpe材料中sv的概念证明,该研究将hi-c方案(即邻近连接测定的全基因组变体)应用于15个ffpe肿瘤样本。在大多数情况下,这种方法(称为“fix-c”)在先前通过fish标记为重排的基因中给出了目测明显改变的接触频率
21
。虽然这种全基因组分析可能与识别新的重排基因相关,但需要昂贵的深度测序,这与需要识别具有已知临床意义的选定基因中重排的临床环境不太相关。
[0474]
在此,我们介绍ffpe靶向基因座捕获(ffpe-tlc),它使用交联dna片段的原位连接,结合寡核苷酸探针组,选择性地下拉、测序和分析具有已知临床意义的基因的邻近连接产物。将ffpe-tlc盲目应用于149个淋巴瘤和对照ffpe样本,通过切除或穿刺活检获得。重排使用“plier”(基于邻近连接的重排识别)自动评分,这是一个专门的计算和统计框架,处理ffpe-tlc测序数据集,并基于其显著富集的邻近连接产物识别目标基因的重排伴侣。fish、靶向ngs捕获和ffpe-tlc结果的比较表明,ffpe-tlc在特异性、灵敏度和检测到的重排的细节方面优于另两种方法。因此,ffpe-tlc是在恶性淋巴瘤和其他易位介导的恶性肿瘤ffpe样本中检测sv的有力新工具。
[0475]
简言之,在ffpe-tlc中,代表性肿瘤样品的ffpe卷轴脱蜡,并轻度去交联,以通过限制性酶(nlaiii)实现原位dna消化,产生中值大小为141bp的片段。原位连接和反向交联后,遵循(基于探针的)杂交捕获的标准方案(详见方法),并在illumina测序机中对所得文库进行测序(图8a和图13)。在我们目前的淋巴瘤探针组中,我们靶向bcl2、bcl6、myc基因和免疫球蛋白基因座igh、igk、igl以及与血液淋巴恶性肿瘤相关的其他基因座。我们将ffpe-tlc应用于129个淋巴瘤肿瘤样本,这些样本选择了是否存在涉及myc、bcl2或bcl6的重排,如fish最初检测到的(图13)。此外,还包括20个来自反应性淋巴结(大多数来自乳腺癌患者)的ffpe样本,这些样本未经fish分析,但预计六个靶基因中没有重排。样本由荷兰五个不同的医疗中心提供,在组织块寿命、dna片段化程度以及坏死和/或挤压损伤的存在方面存在差异(数据未显示)。所有149个样本都是匿名的,因此,在这项(盲)研究中,我们无法得知任何靶基因中是否存在重排。为了说明结果,图8b显示了从典型的ffpe-tlc实验中检索
到的序列的全基因组覆盖率。对myc、bcl2或bcl6的探针靶向基因座处及周围捕获的序列进行更仔细的检查(图8c)突出了ngs捕获与邻近连接结合用于重排检测的附加价值:ffpe-tlc不仅有效地实现了探针互补基因组序列(蓝色),还强烈富集了侧接序列的数兆个碱基(即,如图8c所示的邻近连接产物,其中myc(粉色)、bcl2(棕色)和bcl6(橙色))。由于靶基因座的重排将它们与新的侧接序列并置,重排的伴侣基因座显示ffpe-tlc中邻近连接序列的密度增加,因此可以被发现。这一现象如图8b所示,其中myc(绿色)与含有grhpr基因(红色)的基因座形成异常大量的邻近连接产物,表明肿瘤细胞携带这种易位。
22
.
[0476]
为了以自动化方式客观识别ffpe-tlc数据集中的重排伴侣基因,我们开发了一种称为plier(基于邻近联接的重排识别)的计算流程(pipeline)。简而言之,plier最初将经测序的ffpe-tlc样本解编为多个ffpe-tlc数据集,其中每个数据集由特定靶向基因(例如myc)捕获的邻近连接产物组成。然后,对于给定的(目标基因的)ffpe-tlc数据集,plier评估基因组中的邻近连接产物的密度,以分配和比较基因组间隔的观测和预期邻近评分,并计算富集评分(详见方法和图15)。富集评分显著升高的基因组间隔是靶基因的主要候选重排伴侣。我们最初通过综合优化程序确定了plier的最佳参数(有关优化程序的详细信息,请参阅方法)。然后,我们将plier应用于所有149个样本,以搜索涉及三个临床相关靶基因myc、bcl2和bcl6的重排。图13提供了识别的重排及其与fish诊断比较的概述。在20个对照样本中,ffpe-tlc未检测到重排,证明了plier在屏蔽(ffpe)邻近连接数据集中不可避免存在的固有拓扑和方法学噪声方面的强大能力,且同时能够检测淋巴瘤样本中涉及myc、bcl2和bcl6的重排。
[0477]
总的来说,plier鉴定了137个涉及myc、bcl2和blc6的重排:56个myc重排(49个淋巴瘤样本)、39个bcl2重排(34个样本)和42个bcl6重排(40个样本)(图9a)。为了明确评估plier鉴定的基因组区域是否是所调查靶基因的真正重排,我们在所谓的蝴蝶图中,沿着每个假定伴侣的线性序列仔细检查了它们的邻近连接产物分布
23
。如果参与相互易位,每个基因座应显示一个“断裂点”位置,将优先与伴侣基因座一侧形成邻近连接产物的上游序列与优先接触和连接伴侣基因座另一部分的下游序列分开(图9b)。图9c显示了蝴蝶图所揭示的三个相互重排的例子,分别涉及myc、bcl2和bcl6。重排也可以是非相互的,这样一个目标基因座只有一部分与给定伴侣融合。图9d显示了myc、bcl2和bcl6的这些更复杂重排的蝴蝶图。在所有分析样本中,发现myc涉及41个相互易位(26个igh,15个非ig基因座)和15个更复杂的重排(4个igh),bcl2涉及34个相互易位(33个igh,1个igk)和5个更复杂的重排,bcl6则涉及37个相互易位(16个igh,5个igl,16个非ig基因座)和5个更复杂的重排。
[0478]
除了myc、bcl2或blc6基因座中具有断裂点的137个重排外,plier还有望检测到两种旁观者(bystander)类型的基因组重排,这两种重排也可以在邻近连接产物中产生显著富集。第一个是扩增的基因组区域(拷贝数变异);它们可以与真正阳性重排区分开来,因为plier用所有靶基因对它们进行了评分(图9e)。plier在所有分析的淋巴瘤样本中在整个基因组中发现了23个扩增。plier评分的第二个旁观者类别是基因组重排,涉及包含靶基因的染色体,但在探针靶区外具有断裂点。因此,这种重排在蝴蝶图中所识别的重排和目标位点之间的邻近连接信号中没有线性转变(见图9b)。发现了其中六种重排,对于两种情况(f209和f262),我们确认了涉及第3号染色体的重排,但具有远离bcl6基因座有数兆碱基的断裂点(图16)。plier评分的旁观者重排被认为与感兴趣基因无关,因此被归为阴性。
[0479]
图10a使用circos图
24
提供了本研究中确定重排伴侣的概况图。在我们的样本集合中,发现3个样本在myc、bcl2和bcl6中易位阳性(即三重命中),19个样本在myc和bcl2或bcl6中易位阳性(双命中),8个样本在bcl2和bcl6中重排。在5个肿瘤中,myc或直接与bcl6(f72、f190、f194)基因座融合,要么参与与igh和blc2的复杂三向融合(f197、f274)。除了免疫球蛋白基因座,我们还发现了其他几个重复的重排伴侣,包括kynu/tex41基因座(f67,f188,与bcl6,以及f201与myc),tbl1xr1(f49,f273,f329,与bcl6),ikzf1(f210,f281,与bcl66)和tox基因座(f74,f271,与myc)。引人注目的是,grhpr作为bcl6(f77、f199)和myc(f202、f209、f269)的重排伴侣被发现了5次(图10a)。在f197(myc)和f331(bcl6)等病例中,我们发现了将靶基因座的不同部分融合到不同基因组伴侣的非相互易位事件的强烈迹象(图10b)。在其他情况下,有证据表明等位基因三向重排,通常涉及igh基因座、myc(f50、f212、f274)、bcl2(f193、f274、f282)或bcl6(f77)以及第三个伴侣(例如图10c)。此外,在罕见的情况下,如f67(bcl6)(图10d)、f202(myc)和f197(bcl2),靶基因座的两个等位基因似乎独立地参与了重排。
[0480]
使用ffpe-tlc和plier,我们很容易检索到涉及bcl2、bcl6或myc的137个已识别sv的90个跨断裂点融合读取。将断裂点映射到靶基因以及igh基因座能够检查myc、blc2、bcl6和igh中的复现性断裂点簇,如前所述
5,25
(图10e和图15)。
[0481]
尽管ig基因座的探针设计不是最佳的(因为探针仅集中在增强子区域),但当靶向ig基因时,plier识别出大多数(91个中的79个)与myc、bcl2和bcl6的相互重排。此外,发现许多重排将ig基因座与其他基因连接起来,其中大多数被描述为重排伴侣:igh-pax5/grhpr(f21)
22,26
igh-foxp1(f41)
27
,igh-prdm6(f43),igh-cpt1a(f58)
28
,igl-bach2(f223)
29
和igh-acsf3(f278)
30
。这类病例需要进一步调查,特别是因为它们在未携带其他已知的淋巴瘤驱动因素的样本中发现。
[0482]
为了验证和探索替代的邻近连接方法,我们用4c-seq处理了47个ffpe样品
31
。在4c-seq中,使用反向pcr代替杂交捕获来富集与所选感兴趣位点形成的邻近连接产物
32
。在这项研究中,使用多重4c pcr,14个引物组分布在myc、bcl2和bcl6基因座上,7个引物组针对igh、igl和igk基因座(共21个引物组)。plier的修正版本用于支持ffpe-4c类型的数据和评分重排伴侣(参见方法)。在所有测试样本中,ffpe-tlc和ffpe-4c的结果一致,只有两个例外(f54和f67),其中ffpe-4c未能检测到重排。这两个样本都是2007年和2009年的旧样本,dna严重片段化。这表明,与ffpe-4c相比,ffpe-tlc对较差的样品质量更具耐受性,鉴于4c还需要(小)邻近连接产物的环化,这是可以预期的。
[0483]
我们研究的主要目的是比较ffpe-tlc和fish作为ffpe标本中重排检测的诊断方法。鉴于阴性对照组织中的背景评分结果,如果在少于10-20%的细胞中出现异常信号(每个基因和每个诊断中心的确切截留值可能不同),则在诊断实践中通常认为fish为阴性。ffpe-tlc的灵敏度取决于plier识别候选重排伴侣的能力。为了更系统地研究plier性能和灵敏度,我们取了六个ffpe样品,这些样品在myc(2x)、bcl2(2x)和bcl6(2x)中携带fish验证的重排,fish阳性细胞的百分比已知,用未携带重排的对照物稀释每个样品(在探针下拉之前)至5%、1%和0.2%的百分比。我们发现,plier在任何样本中都没有发出假阳性调用,并可信地为所有具有5%或更多阳性细胞的样本中的实际重排伴侣评分(见图11a-b和图17)。这表明,与fish相比,ffpe-tlc具有更高灵敏度。然而,低肿瘤细胞百分比或肿瘤异质
性导致的低易位百分比的临床意义有待确定。
[0484]
我们将原始fish结果与我们的ffpe-tlc结果进行了比较。在通过ffpe-tlc评分为myc阳性的49个样本中,有47个样本也通过fish进行了分类(图13)。fish忽略的myc重排都是顺式的,伴侣位于同一条8号染色体上(f16和f221:f此处fish检测到多个信号)(图11c)。对于bcl2,我们评分为阳性的34个样本中,有31个之前也曾被fish报告过:三个新发现的重排,每个都携带bcl2-igh易位,尚未被fish分析。对于bcl6,40个bcl6重排肿瘤中的29个也通过fish进行了评分。fish未检测到三种bcl6重排(f38、f40、f49)(图11d),在其中两种情况下,因为重排细胞的百分比低于阈值(10%(f38)和6%(f40))。在第三种情况(f49)中,ffpe-tlc检测到tbl1xr1基因座在bcl6基因座中插入1.35mb(图11e)。事后看来,在fish图像(图11f)中可以观察到一些信号的分裂,这最初被认为是不相关的。由于单个荧光信号(f25、f261),fish先前认为两个ffpe-tlc鉴定的bcl6重排(其中一个是与igh)是不确定的。fish未分析过六个新发现的bcl6重排(2x igh,2x igl)(图13)。相反,fish评分的所有重排均由ffpe-tlc确认,除了两个(f217和f322,均被描述为具有复杂的核型)。不幸的是,这里无法确定ffpe-tlc或fish是否错误。总之,所有分析的149个样本ffpe-tlc显示出与fish的高度一致性。它错过了两个由fish评分为明显的重排,但也鉴定并表征了fish未评分的两个myc重排和五个bcl6重排。此外,ffpe-tlc能够并行分析多个基因参与重排的能力,使其能够在未通过fish测试这些重排的样本中发现9例bcl2和bcl6重排。在四个案例中,这一发现改变了样本的原始肿瘤分类。样本f16从myc和bcl2重排的“无命中”重新分类为“双命中”(dh),样本f67从单次(myc)命中重新分类为myc-bcl6 dh肿瘤(与伴侣igh和igl),样本f194从单次(myc)命中重新分类至myc-bcl2-bcl6三重命中(th,尽管myc和bc l6融合在一起),样本f209从dh重新分类为th。
[0485]
我们还希望将ffpe-tlc与基于dna捕获的靶向测序方法(capture-ngs)进行比较,以检测和分析ffpe样本中的结构变体
5-7
。为此,我们比较了capture-ngs和ffpe-tlc在19个ffpe样本上的性能,这些样本是capture-ngs之前分析的》200个ffpe样本的更大队列的一部分。所选样本包括一个子集,其中capture-ngs结果与原始fish诊断不一致。图12a显示了该比较的结果。六个ffpe淋巴瘤样本中的全部六个样本(其中capture-ngs未能识别总共七个fish报告的易位)被ffpe-tlc确认携带七个报告的易位(样本f190(myc和bcl6)、f197和f198(myc)、f193(bcl2)、f188、f191和f192(均为bcl6))。为了揭示capture-ngs错过这些重排的潜在原因,我们发现在三种情况下,实际断裂点位于capture-ngs探针靶向区域之外(f188、f197、f192)。在一个病例(f190)中,ffpe-tlc证明fish鉴定的myc和bcl6重排实际上是单个myc-bcl6易位。capture-ngs未能找到断裂点融合读取,因此错过了该重排,因为bcl6断裂点位于探针靶向区域之外,而myc断裂点位于探针无法覆盖的重复序列中(图12b)。因此,在断裂点发生在探针覆盖区域之外的情况下,capture-ngs无法识别重排,而ffpe-tlc如讨论的,检测这种重排没有问题。为了进一步说明这一点,我们重新分析了六个样本的数据集,这些样本携带fish确认的bcl2(2x)、bcl6(2x)或myc(2x)重排,但对读取进行了过滤,以专门考虑距离映射断裂点以50kb间隔越来越远的捕获:在所有情况下,plier都以非常高的置信度发现了重排(图12c)。在其他三种情况下(f191、f192、f198),capture-ngs无法识别重排伴侣,因为它在非独特序列上断裂并融合。为了进一步评估ngs策略在基于断裂点融合读取映射识别重排时可能存在的困难,我们分析了本研究中发现的所有断裂
点侧接序列在不同读取长度上的可映射性。图12d显示,约5%的已识别重排不可独特映射,因此即使在将50个核苷酸读入伴侣序列时也会丢失。相反,有一例capture-ngs识别的融合读取表明myc易位,fish和myc免疫组化未证实,ffpe-tlc也未对易位进行评分(f189)。通过pcr和测序进行的进一步详细分析表明,这是一个小插入,将8号染色体的240个碱基对插入x染色体,但不影响myc基因座(图12e)。
[0486]
总之,ffpe-tlc在检测染色体重排方面优于常规capture-ngs方法。capture-ngs依赖于断裂点融合读取识别来检测重排,当断裂发生在探针覆盖区域外和/或重复dna中时,会受到严重阻碍。如我们所示,ffpe-tlc准确地发现了这些重排,因为它分析了靶基因与其重排伴侣之间的邻近连接对。
[0487]
讨论
[0488]
我们在此介绍ffpe-tlc,这是一种基于邻近连接的方法,用于靶向鉴定ffpe肿瘤样本中临床相关基因的染色体重排。作为一种应用于诊断环境的检测方法,ffpe-tlc与fish相比具有重要优势,fish是目前淋巴瘤ffpe样本中靶向重排检测的金标准。首先,与ffpe-tlc不同,fish高度依赖于高质量的组织和细胞形态,这可能会受到切除标本中的坏死、凋亡和挤压伪影以及来自芯针活检样本材料非常有限的负面影响。我们在这项研究中包括了芯针活检样本,这表明即使是非常小的样本也能获得高质量的ffpe-tlc结果。第二,fish结果可能给出不确定的结果,或者在每个细胞中见到异常数量fish信号的情况下导致主观解释;ffpe-tlc提供了基于数据分析算法plier对涉及所选靶基因位点重排进行客观评分的巨大益处。第三,ffpe-tlc结果提供了更详细的重排信息:该方法不仅同fish一样对临床相关基因是否完整或经重排进行评分,还识别了重排伴侣、断裂相对于相关基因的位置,以及通常以碱基对分辨率描述重排的融合读取。收集与疾病进展和治疗反应相关的详细信息有望改善癌症患者的诊断、预后和治疗。碱基对水平上的易位信息也提供了个体化的肿瘤标志物,从而能够设计用于最小残留疾病检测的肿瘤特异的个体化分析。最后,ffpe-tlc更灵敏:为了避免假阳性调用,fish评估通常使用10-20%的异常信号截留点,如正常对照参考所设置的,这些异常信号是由于3-5μm切片中直径10-20μm的肿瘤细胞的“截留”信号引起的。即使只有5%的细胞存在ffpe-tlc也能可靠检测重排,这使得它成为一种应用于实体瘤融合基因检测的有趣方法。
[0489]
常规ngs-捕获方法也用于识别sv,寻找融合伴侣并提供重排断裂点的详细信息,但与这些方法相比,ffpe-tlc具有重要优势,特别是因为它不严格依赖于成功下拉和融合读取的识别。相反,ffpe-tlc测量断裂点两侧染色体间隔之间聚集的邻近连接事件,以识别重排。正如我们所示,这使得能够对常规ngs-捕获方法遗漏的重排进行强力检测,例如,当探针定位靠断裂点不够近以拉低融合读取时,或者当断裂点两侧的非独特序列对融合读取识别有害时。
[0490]
我们研究的一个关键方面是plier的开发,plier是我们的计算/统计流程,用于客观调查ffpe-tlc数据集的重排伴侣。目前使用的处理靶向-ngs法产生数据的融合读取查询器通常需要一定程度的手动数据管理,从而阻止了完全自动化和并行数据处理。在ffpe-tlc中,plier实现了染色体重排的自动识别,从处理经测序ffpe-tlc文库到提供包括已识别重排的简单表格。plier在每个测试样本中搜索具有显著富集的独立连接片段密度的染色体间隔,而无需与参考(或对照)数据集进行比较。因此,它考虑了样本之间固有信号噪声
水平差异,这是至关重要的,因为来自不同组织、不同医院和不同档案存储时间和条件的ffpe样本的dna质量范围相对较大。最初在6个样本的精选数据集上进行训练,然后应用于所有样本的完整数据集,plier证明对不同水平的噪声非常强,同时对检测我们研究中所有149个样本的重排非常灵敏。
[0491]
根据世界卫生组织(who)对淋巴瘤的分类,本研究中发现的恶性淋巴瘤的大量重排值得考虑。目前,具有myc-和bcl2和/或bcl6联合易位的侵袭性b细胞淋巴瘤(所谓的双命中或三命中,dh/th淋巴瘤)被分类为一个单独实体,而不考虑形态学特征。这一点的基本原理不仅体现在“有生物学意义的分类”的目标上,还体现在特征性的不良临床结果上,这证明了加强一线治疗的合理性。最近,在一大系列的此类淋巴瘤中,lunenburg淋巴瘤生物标志物联盟可能表明,这种不良结果实际上仅限于具有myc重排的ig伴侣的dh/th淋巴瘤,而所有其他情况(myc单命中,非ig伴侣)具有与没有myc重排的dlbcl相似的结果。因此,在不久的将来,病理学家将需要在这样的水平详细提供侵袭性b细胞淋巴瘤的易位状态,以支持治疗决策。使用fish,需要4种单独的检测(bcl2、-ba(断裂-分离)、bcl6-ba、myc-ba、myc-igh-f(融合))来诊断dh/th淋巴瘤,但仍然缺少携带myc-igl易位的病例,因为没有商业探针可用于myc-igl融合fish。使用ffpe-tlc,也可以在一次检测中可靠诊断这种易位情况,这明显提高了时间和成本效益。我们鉴定了4例myc-igl和1例myc-igk患者,其中1例dh患者(f264)的临床后果是立竿见影的。我们注意到三例myc-bcl6融合(f072、f190、f194)和两例myc、bcl2和igh融合(f197、f274)的病例通过fish无法识别,并在四例病例中解释为dh,在一例中解释为th。然而,尚不清楚单个易位事件是否活化了两个易位伴侣基因,并导致了与两个独立事件类似的生物学影响。类似地,myc和bcl6都经常易位到对恶性b细胞行为具有可能生物学影响的基因(例如tbl1xr1、ciita、ikzf1、mef2c、tcl1)。然而,到目前为止,还无法在临床环境中研究这种融合伴侣的影响。
[0492]
总之,ffpe-tlc结合plier进行客观重排调用比常规ngs-捕获方法和fish在淋巴瘤ffpe标本的分子诊断方面具有明显优势。未来的前瞻性研究应证明ffpe-tlc在其他癌症类型,如软组织肉瘤、前列腺癌和非小细胞肺癌(nsclc)中的表现,它们也经常携带临床相关的染色体重排。
[0493]
参考文献
[0494]
1.-m
á
rmol,a.m.等人,myc status determination in aggressive b-cell lymphoma:the impact of fish probe selection.histopathology 63,418-424(2013).
[0495]
2.scott,d.w.等人,high-grade b-cell lymphoma with myc and bcl2 and/or bcl6 rearrangements with diffuse large b-cell lymphoma morphology.blood 131,2060-2064(2018).
[0496]
3.copie-bergman,c.等人,myc-ig rearrangements are negative predictors of survival in dlbcl patients treated with immunochemotherapy:a gela/lysa study.blood 126,2466-2474(2015).
[0497]
4.cassidy,d.p.等人,comparison between integrated genomic dna/rna profiling and fluorescence in situ hybridization in the detection of myc,bcl-2,and bcl-6gene rearrangements in large b-cell lymphomas.american journal of clinical pathology 153,353-359(2020).
with bwa-mem.arxiv preprint arxiv:1303.3997(2013).
[0527]
34.geeven,g.,teunissen,h.,de laat,w.&de wit,e.peakc:a flexible,non-parametric peak calling package for 4c and capture-c data.nucleic acids research 46,e91-e91(2018).
[0528]
35.collette,a.python and hdf5:unlocking scientific data.("o'reilly media,inc.",2013).
[0529]
36.de ridder,j.,uren,a.,kool,j.,reinders,m.&wessels,l.detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens.plos computational biology 2,e166(2006).
[0530]
材料和方法
[0531]
患者样本:这项回顾性研究使用了一组129个存档的b细胞非霍奇金淋巴瘤组织样本,这些样本是由各个位点选择的,因此可能不代表各个位点样本的完全随机选择。2007年至2019年间,在乌得勒支大学医学中心、阿姆斯特丹大学医学中心(vumc)、荷兰病理学实验室、莱顿大学医学中心和格罗宁根大学医学中心及其附属医院诊断出相应的淋巴瘤患者。他们大多被诊断为dlbcl,但也包括burkitt、滤泡和边缘区淋巴瘤以及其他一些诊断。还分析了20个非淋巴瘤对照样本,主要是反应性淋巴结样本和扁桃体切除术样本。使用标准诊断程序获得福尔马林固定和石蜡包埋(ffpe)组织样本。每个患者提供1个或多个10μm卷轴或4μm未染色的ffpe组织块切片,用于试管内或载玻片上的ffpe-tlc分析。本研究是根据当地机构委员会的要求进行的,在本研究期间遵守了所有相关的道德和隐私法规。
[0532]
分子分析:在选定的病例中,所有患者样本都用常规fish进行了分析,使用分离探针和融合探针,大部分情况下,分析了所有3个基因bcl2(cytocell lps028;vysis abbott 05n51-020;igh/bcl2 dual fusion vysis abott 05j71-001),bcl6(细胞lph 035;vysis abbott 01n23-020)和myc(细胞lps 027;vlysis abbott 05j91-001;igh/myc/cep 8dual fusion vysis abott 04n10-020)。还用阿姆斯特丹大学医学中心vumc团队开发的capture-ngs方法对19个样本的子集进行了分析。补充材料和方法中提供了该方法的详细说明。
[0533]
ffpe-itc文库制备:简言之,本研究中医疗中心提供的单个ffpe切片为1.5ml小瓶或载玻片中的卷轴。如果提供载玻片,则刮下载玻片中所含物质并将其转移到1.5ml小瓶中。通过3分钟80℃热处理去除过量石蜡,然后进行离心步骤,然后使用m220聚焦超声发生器(covaris)通过超声破碎组织并匀浆。通过在80℃下与0.3%sds孵育2小时引发样本用于酶消化,然后在37℃下用nlaiii(一种4bp切割酶;neb)消化1小时,最后在室温下用t4 dna连接酶(roche)连接2小时。接下来,通过在80℃下孵育过夜完全逆转交联,并使用异丙醇沉淀和磁珠分离纯化dna。洗脱后,将100ng制备的材料片段化至200-300bp(m220聚焦超声发生器,covaris),并进行ngs文库制备(roche kapa hyperprep,kapa独特双索引衔接子试剂盒)。将总共16-20个独立制备的文库等摩尔合并,总质量为2μg,并根据制造商的说明使用roche hypercap试剂和工作流程与捕获探针库杂交,洗涤和pcr扩增。在illumina novaseq 6000测序机上进行配对末端测序。所有邻近连接文库的测序都比认为必要的更深入。具有最低覆盖率的样本测序到大约20m的读取深度,这总是足以进行重排检测。
[0534]
ffpe-tlc数据处理:使用bwa-mem(设置:-sp-k12-a2-b3)在配对末端模式下将单
个样本(即患者)的序列读取映射到人基因组(hg19)
33
。bwa-mem比对仪允许“分裂-映射”,其中单个读取可以映射到基因组中的多个片段(即分离区域)。这对于映射ffpe-tlc数据至关重要,因为ffpe-tlc中的每个测序读取可能包含映射到基因组中不同位置的多个片段(见图14)。具有0以上的映射质量(mq)的任何片段都被认为是映射的,这通常用于邻近连接数据处理
32,34
。根据片段与视点坐标的重叠,将读取分配给相关靶基因或“视点”(即myc、bcl2等探针集)(探针集坐标见图18)。如果读取的内容与任何视点都不重叠,则会将其丢弃。如果读取中的片段与多个视点重叠,则将读取分配给重叠最大的视点。作为该过程的结果,对于样本和视点的每个组合都会生成一个独立的ffpe-tlc比对文件(bam)。
[0535]
根据nlaiii限制性酶(catg)的识别序列,将参考基因组在计算机上拆分为“区段”,其中每个区段以nlaiii识别位点开始和结束。然后将映射片段覆盖在片段上。由于罕见的比对错误,一次读取中的多个片段可能会与一个区段重叠。在这种情况下,该特定区段仅算一个片段,并且忽略了该读取上额外重叠片段。我们使用hdf5格式
35
存储ffpe-tlc数据集,这是一种跨平台和跨语言的文件存储标准,因此为ffpe-tlc的未来用户提供了便利。
[0536]
重排识别:见de ridder等人
36
,其目的是鉴定整个基因组中信号(即覆盖)超过预期的富集。在给定ffpe-tlc数据集中,plier最初将参考基因组分割成等间距的基因组区间(例如5kb或75kb的箱),然后计算每个区间的“邻近频率”,该频率定义为该基因组区间内被至少一个片段(即接近连接产物)覆盖的区段数量,有关整个过程的示意图,请参见图6。然后,通过对每个染色体上的邻近频率进行高斯平滑来计算“邻近评分”,以消除最可能是虚假的邻近频率非常局部和突然的增加(或减少)。接着,通过对整个基因组中观察到的邻近频率进行计算机内改组,然后跨越每个染色体进行高斯平滑,来估计具有相似性质的基因组间隔(例如,跨染色体存在的基因组间隔)的预期(或平均)邻近评分和相应的标准偏差。最后,使用其观测邻近评分和相关的预期邻近评分及其标准偏差来计算每个基因组间隔的z-得分。最后,通过组合从多个规模(即间隔宽度,如5kb和75kb)计算的z-得分,计算规模不变的富集评分(详见plier部分的富集评分估计和参数优化)。该规模不变的富集评分用于识别具有观测连接产物的升高聚类的基因组间隔。
[0537]
对于顺式染色体上存在的基因组间隔,我们首先校正了与目标基因座相邻的基因组间隔已知升高的邻近频率。为此,对于给定的ffpe-tlc数据集,我们最初排除了探测区域以及周围+/-250kb的区域。然后,我们对探测区域两侧的邻近频率进行高斯平滑(σ=0.75,跨度=31个间隔),直到染色体终止。接下来,受峰c
34
的启发,我们对平滑的邻近频率进行了保序回归。对于每个顺式间隔,我们将其平滑的邻近频率和对应的保序回归预测值之间的差异视为其邻近评分。该程序确保了与目标(或探测的)基因座相邻的基因组间隔中邻近评分的已知升高被考虑在内。最后,顺式间隔的富集评分按照与反式间隔类似的改组程序计算(如上所述)。我们丢弃了在视点周围+/-3mb区域中识别的顺式重排(即,在线性染色体上测定距离视点小于3mb),以确保视点之间及其附近的真实3d相互作用不被视为重排。
[0538]
值得注意的是,当ffpe-tlc数据集不稀疏,并且至少最小程度上使用独立连接产物(即基因组中不同基因组片段的覆盖率)时,上述统计方法工作良好。然而,稀疏的ffpe-tlc可能产生于以较差样品(组织)质量、dna提取、低消化或连接效率或文库制备中的其他困难制备的文库。在这种情况下,基因组中只有最小数量的基因组间隔具有高于零的邻近评分。结果,所使用的置换策略(即间隔的随机改组)将低估真实的预期邻近评分,因此许多
邻近评分高于零的间隔将被错误地认为是富集的。为了解决这一问题,我们考虑了一种互补置换方法,其中我们仅交换邻近频率高于零的基因组间隔(而不是所有间隔的随机改组),然后通过比较使用交换置换策略计算的观测和预期邻近评分来计算相应的z-得分。对于每个基因组间隔,我们将改组和交换置换之间的最小z-得分作为特定基因组间隔的最终z-得分。即使在稀疏的ffpe-tlc数据集中,这一增加也限制了假阳性调用的数量,并使plier也适用于ffpe-4c实验。在所有置换中,我们重复改组或交换1000次,以估计邻近评分的相应预期和标准偏差。
[0539]
需要注意的是,在这种方法中,我们没有纠正已知的偏差,例如gc含量、可映射性、区段或限制位点密度(即每个间隔的限制性位点数量)或可能影响捕获的邻近频率的许多其他已知因素。由于plier的灵活性,可以通过仅交换(或改组)具有相似染色质区室、gc含量、限制性位点密度等的间隔来在背景估计中考虑这些参数。尽管如此,当在背景估计中校正这些参数时,我们的最初分析没有显示出显著改善,因此我们选择了简单的模型,这反而降低了plier的计算需求。这一决定尤为重要,因为我们的目标是生产一种适用于临床环境、计算需求最低的轻量级流程。plier的源代码可从github下载,网址为https://github.com/delaatlab/plier.
[0540]
富集评分估计:对于给定样本(例如患者)和视点(例如bcl2)和基因组间隔宽度(例如5kb),我们最初选择了z-得分高于5.0的基因组间隔,如果它们接近1mb,则合并相邻的所选间隔。我们将合并间隔的90%z-得分作为其综合z-得分。为了从多个间隔宽度(例如5kb和75kb)估计“规模不变”的富集评分,我们对接近10mb的合并间隔进行分组,并将具有最大规模的间隔(在此为75kb)的z-得分作为最终富集评分。在本研究中,跨规模的每个合并间隔集合被称为一个“调用”。
[0541]
plier的参数优化(即训练阶段):为了鉴定plier的最佳参数,我们使用了六个ffpe-tlc样本、三个淋巴瘤(“阳性”)和三个对照(“阴性”)样本的集合。具体而言,包括了三个淋巴瘤样本(即f73、f37和f50),根据fish(金标准),这些样本预计分别在bcl2、bcl6或myc中具有单个重排,而在另两个基因中缺乏重排。其他三个“阴性”数据集(即f29、f30和f33)是对照数据集,预期三个基因中的任何一个都没有重排。我们将优化限于bcl2、bcl6和myc基因,因为我们只有这些基因的临床/诊断fish数据。我们还将三个淋巴瘤样本(即f73、f37和f50)的稀释(即5%、1%和0.2%)实验纳入优化程序。综合起来,我们有12例阳性病例(3名原始患者,加上每名患者的3个额外稀释样本),plier应识别出其中的重排(即“真阳性”集合),33例阴性病例(3个对照样本每个有3个基因,加上12个淋巴瘤样本中的两个未重排基因),plier不应识别出基因组中的任何重排(即。“真阴性”集合)。除了正确识别的重排外,在阳性病例的整个基因组中发现的任何额外重排也被认为是“假阳性”重排。作为性能测量,我们使用精确召回率下面积(auc-pr)代替曲线下面积,因为我们可能有更多的负面例而不是正面例(即不平衡分类频率)。
[0542]
为了有效执行plier的统计框架,需要优化确定几个参数。我们使用乌得勒支大学医学中心的高性能计算(hpc)进行了大规模参数扫描,以确定plier的最佳参数。这些参数包括:高斯平滑程度(σ=0.1、0.25、0.5、0.75、1.0、1.5、2.0、2.5、3.0、3.5、4.0)、高斯内核跨度的基因组间隔数(#步长=11、21、31、41、51、61)和基因组间隔宽度(宽度=5kb、10kb、25kb、50kb、62kb、75kb、100kb)。对于间隔宽度,我们还测试了组合多个间隔宽度(即规模不
变的富集评分)是否表现更好。此外,为了确定合并间隔(即彼此相邻1mb内的间隔)的z-得分应如何整合,我们考虑使用最大、90%和中值运算符进行实验。
[0543]
在参数扫描后,我们确定以下为plier的最佳参数:高斯平滑σ=0.75,高斯内核跨度#步长=31,间隔宽度=5kb+75kb(即两个z-得分都应高于5.0),相邻区间(《1mb)间隔的90%的z-得分合并为其最终z-得分。最后,需要估计显著性阈值,以考虑显著富集的调用。通过将最大错误发现率(fdr)设置为1%,我们达到了8.0的显著性,作为反式间隔富集评分的最佳显著性阈值。由于计算限制和诊断数据的可用性有限,我们仅优化了bcl2、bcl6和myc反式间隔的plier参数。然后,我们使用这些参数(不经进一步优化)用于研究中其他基因(即igh、igl和igk)的反式间隔。对于我们研究中所有基因的顺式间隔,我们再次使用了上述参数,但显著性阈值除外。对于这些调用,我们采用了更高显著性阈值(即》16.0)的保守方法。plier的每个输出调用由两个基因组坐标组成,其指示规模不变富集评分高于显著性阈值的边界。
[0544]
扩增检测:尽管ffpe-tlc并非设计用于识别扩增,但plier从不同探针组但在相同样本和区域中识别的重复重排可以指示该区域中的扩增事件。为了充分利用这一前景,我们将重点放在研究中的三个主要基因(即myc、bcl2和bcl6)上,对其进行了相对较大区域的研究(详见图18)。对于每个样本,我们询问是否从多个基因中报告了特定重排(即在同一区域中)。图9e描述了plier识别的此类扩增的例子。值得注意的是,淋巴瘤样本可能携带对igh区域特异性的双命中重排(例如bcl2和myc)。为了避免将这种重排调用为扩增事件,我们从扩增检测分析中排除了对igh区域的调用。
[0545]
黑名单区域:我们注意到,我们的igl和igk探针组倾向于重复识别基因组中的特定区域。即使在我们的对照样本中,我们也观察到了这样调用,因为其预期不会出现重排。具体而言,我们的igl探针组频繁鉴定了chr9:131.5-132.5mb,而我们的igk探针组频繁鉴定了人(hg19)基因组的chr22:22-24mb区域。值得注意的是,chr22:22-24mb区域携带igl基因,因此此类调用可能值得进一步研究。然而,我们注意到对应的igl视点并不能相互识别igk。因此,我们认为富集评分升高是由于igl和igk之间的高度序列相似性,这可能导致映射过程中的错误比对。总之,我们分别将这两个区域视为igk和igl探针的脱靶结合,并忽略了这两个探针组在这些区域中识别的任何重排。
[0546]
融合读取识别:为了识别给定ffpe-tlc数据集(例如myc)中的融合读取,我们收集了分裂-比对(即映射到基因组中多个区域的单个读取序列)。然后,通过丢弃在基因组中限制性酶识别位点(+/-1碱基对)处融合的分裂-比对,筛选出ffpe-tlc中涉及酶消化的分裂-比对。在igv中手动检查重排坐标(由plier识别)处发生的分裂-比对,以确认存在读取融合。
[0547]
融合读取可映射性:从融合读取中识别的断裂点坐标用于可映射性分析,以从参考基因组中提取相应的序列。从参考基因组中提取了断裂点上游和下游总共347个151bp(等于测序读取长度)的序列。这347个序列使用blastn(设置:-perc_identity 80-dust no-evalue 0.1)在20到151的不同序列长度上进行比对,步长为1bp。对blast结果进行分析,以计算每个长度上精确命中的序列;如果恰好一次命中,则序列被认为是独特的,如果多次命中,则该序列被认为不是独特的。非独特序列的比例绘制成柱状图。
[0548]
确认样品f189中240bp的chr8插入chrx:对对照dna和从样品f189(nebnext q5混
合物,neb)中分离的dna进行2x 20轮嵌套pcr,使用两个引物用于chrx插入侧翼的初始pcr(正向:attttgatcggcttagcca,反向:ggttgatcaaagccagtc)pcr,使用两个引物用于嵌套pcr(正向:gtccagctttgtcctgtatt,反向:gtcatggctggtcaagtag)。pcr产物在琼脂糖凝胶上分离,显示仅针对样品f189形成了具有插入的预期大小的产物(数据未显示)为了进一步确认,在相同的嵌套pcr中扩增初级pcr产物,但现在包括illumina测序衔接子和索引序列(正向:gtgactggagttcagacgtgtgctcttccgatctgtccagctttgtcctgtatt,反向:accctttccctacacgacgcttccgatctgtcatggctggctggtcaagatag),并进行测序(illumina miniseq)。
[0549]
数据可用性:本研究中使用的所有测序数据都映射到参考基因组(hg19),并可通过欧洲基因组-表型组数据库获得。
[0550]
补充材料和方法:捕获-ngs
[0551]
dna分离、文库制备和测序:根据制造商的方案,使用qiaamp dna ffpe组织试剂盒(qiagen,hilden,germany)从3-10x 10μm ffpe切片中提取dna。根据制造商的离心方案,使用qiaamp blood mini试剂盒(qiagen,hilden,germany)提取外周血dna。使用qubitbr试剂盒(thermo fisher scientific,carlsbad ca,usa)用qubit 2.0荧光计定量分离的dna,并用covaris s2或me220(covaris inc,woburn ma,usa)对250-800ng(总体积130μl)片段化,covaris s2为200个循环/脉冲串6分钟至平均尺寸180-220bp,和1000个循环/脉冲串3分钟,至平均大小为250-300bp。使用agilent dna 1000试剂盒(agilent technologies,santa clara,ca),用2100生物分析仪测定dna浓度和片段化情况/大小分布。使用250ng的180-220或250-300bp片段化dna,用kapa文库制备试剂盒(kapa biosystems,wilmington ma,usa)创建ngs文库。简而言之,修复dna末端(20℃,30分钟),连接单个a-尾(30℃,30分钟)。随后,将独特索引的衔接子(roche nimblegen,madison wi,usa;idt,coralville ia,usa)连接过夜(16℃),然后进行大小选择以保留250-450bp之间的片段。dna扩增了七个聚合酶链反应(pcr)循环。对创建的dna文库的等分试样进行靶向捕获。使用nimblegen设计软件(roche)设计了捕获组。捕获组包括用于突变分析的约350个基因(约1.5mb)的外显子和用于易位分析的多个染色体区域(包括基因、内含子和基因间区域;约1.5mb)(罗氏订单id 0200204534、id 43712和id 1000002633)。根据nimblegen ez-seqcap文库方案v5.1(roche nimblegen,madison wi,usa)进行捕获。每次捕获时,八个文库的dna在一个试管中等量合并成总共1μg dna。在47℃下探针杂交过夜。合并物扩增14个pcr循环。将三个合并物等摩尔合并并加载到一条序列泳道上,并分别在hiseq 2500或4000上对125bp或150bp配对末端进行测序。
[0552]
序列读取的比对:ngs读取用bcl2fastq(illumina)解编。使用seqpurge(-最小长度20;v0.1-104)修正衔接子和劣质碱基。读取与人类参考基因组(hg19)用bwa mem(-m-r;v0.7.12)比对(heng 2013)。使用abra(v0.96)(mose等人,2014)对读取重新比对以提高比对准确度。使用sambamba(v0.5.6)按查询名称对比对的bamfile进行排序,并使用picardtools markduplicates(v2.4.1)标记重复读取,设置assume sort order=queryname(查询名称)。除了标记重复的主要比对外,还需要此设置来标记重复的次要比对。(tarasov等人,2015;'picard tools').接下来,读取按坐标(sambamba)排序,以与数据分析流程的其余部分兼容。
[0553]
结构变体分析:结构变体分析的流程部分包括易位、反转、删除、插入和重复,是在
工作流管理系统snakemake中创建的(和rahmann 2012)。为了获得高灵敏度和特异性,组合4种易位检测算法:breakmer(v.0.0.4)(abo等人,2015),gridss(v.1.4.2)(cameron等人,2017),novobreak(v.1.1.3)(chong等人,2017)和wham(v.1.7.0)(kronenberg等人,2015)。这些基于以下条件选择:1.检测易位的可能性,2.适用于具有短插入尺寸的配对末端illumina测序数据。3.可在靶向测序数据上使用。4.可建档。5.至少从2017年维持。使用默认设置执行breakmer、gridss和novobreak。wham以映射质量为10(-p),碱基质量为5(-q)执行。为了与breakmer兼容,从bamfile中删除了染色体前缀。breakmer需要包含易位检测感兴趣区域的靶bed文件,为了减少组装时间并获得更高的准确性,易位靶在靶bed文件中划分为5kb的区域。
[0554]
为了能够组合这些工具的输出,将输出转换成r(v.3.4.1)从而能够在工具之间比较,并添加了基因注释。为了除去噪声,添加了过滤器。按顺序从数据中删除以下sv:
[0555]
两个断裂点均脱靶的sv,在捕获探针位置外还超过300bp。
[0556]
使用同一工具检测到具有完全相同断裂点的重复sv。
[0557]
不符合工具预设阈值的sv。对于breakmer,至少有4个分裂读取和3个不一致读取,对于wham,至少有8个读取(不一致和分裂读取的总和),对于gridss,质量分数高于450,对于novobreak,平均覆盖至少4个高映射质量易位读取。
[0558]
组合四个工具的sv输出,仅移除一个工具检测到的sv。因此,仅包括了至少两种工具识别的sv。因此,落在10bp范围内的断裂点被认为是相同的sv。
[0559]
黑名单:检查结果显示多个经常复发的sv。在整合基因组浏览器(igv)中对这些事件的手动检查告诉我们,这些sv是不同来源的人工产物。部分人工sv是基因组中高度重复区域的结果,其他由部分同源区域引入。此外,在数据中还检测到一些常见的种系sv,特别是小的插入缺失。为了从输出中除去这些问题区域,基于25个非肿瘤样本(12个血液样本、4个ffpe增生淋巴结、6个ffpe反应性淋巴结和3个ffpe上皮组织)创建了黑名单。对于这25个样本,sv检测是按照完全相同的dna、分离、制备和测序以及四种选择的检测工具在相同的设置下进行的。使用bed-tools multi-inter(v0.2.17)将至少2个非肿瘤样本中检测到的在10bp范围内常见断裂点位置添加到黑名单中。距离不到50bp的黑名单区域用bedtools合并为一个区域。从sv检测输出中移除在黑名单区域内具有断裂点之一的sv。在igv中手动检查剩余sv。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1