准确检测DNA单分子中突变的方法与流程
发明领域本发明涉及一种生成用于测序的核酸文库的方法及其用途,该核酸文库的错误率为每1亿个碱基对<1个错误。本发明提供了用于检测存在于单个dna分子(例如,来自多克隆的细胞群体的单个dna分子)中的突变(包括驱动突变(driver mutation))的方法,以及该方法在体内或体外测量体细胞突变率、突变特征(mutational signature)和驱动突变中的用途。本发明还提供了发现涉及疾病的体细胞突变和检测来自培养细胞的突变、诊断疾病和检测体外培养和/或经受体外诱变过程的细胞中可疑突变的存在和/或特征(identity)的方法。本发明还还提供了一种双链测序数据的计算分析的方法。
背景技术:
0、发明背景
1、成人组织通常是多克隆的,由遗传上异质的细胞或携带终生积累的不同体细胞突变的细胞克隆组成。研究正常组织中的体细胞诱变需要新的方法来规避这种遗传异质性,例如对小活检组织进行超深度测序以检测小克隆(martincorena等人,(2015)doi:https://doi.org/10.1126/science.aaa6806和martincorena等人,(2018)doi:https://doi.org/10.1126/science.aau3879);显微组织学单位的激光显微切割(lee-six等人,(2019)doi:https://doi.org/10.1038/s41586-019-1672-7和brunner等人,(2019)doi:https://doi.org/10.1038/s41586-019-1670-9);分离单个细胞,随后在体外扩增成克隆类器官或克隆(welch等人,(2012)doi:https://doi.org/10.1016/j.cell.2012.06.023,jager等人,(2017)doi:https://doi.org/10.1038/nprot.2017.111和franco等人,(2018)doi:https://doi.org/10.1038/s41467-018-03244-6);或使用全基因组扩增的直接单细胞测序(lodato等人,(2015)doi:http://doi.org/10.1126/science.aab1785 and lodato等人,(2018)doi:https://doi.org/10.1126/science.aao4426)。虽然这些方法改变了我们对几种组织的突变图的理解,但由于扩增错误,单细胞测序的错误率通常很高,并且所有其他方法通常限于有丝分裂活性细胞类型,其或者排列在组织内的克隆斑块(clonalpatch)中,或者可以在体外从单细胞扩增。因此,大多数人细胞类型中体细胞突变的速率和模式仍然未知。
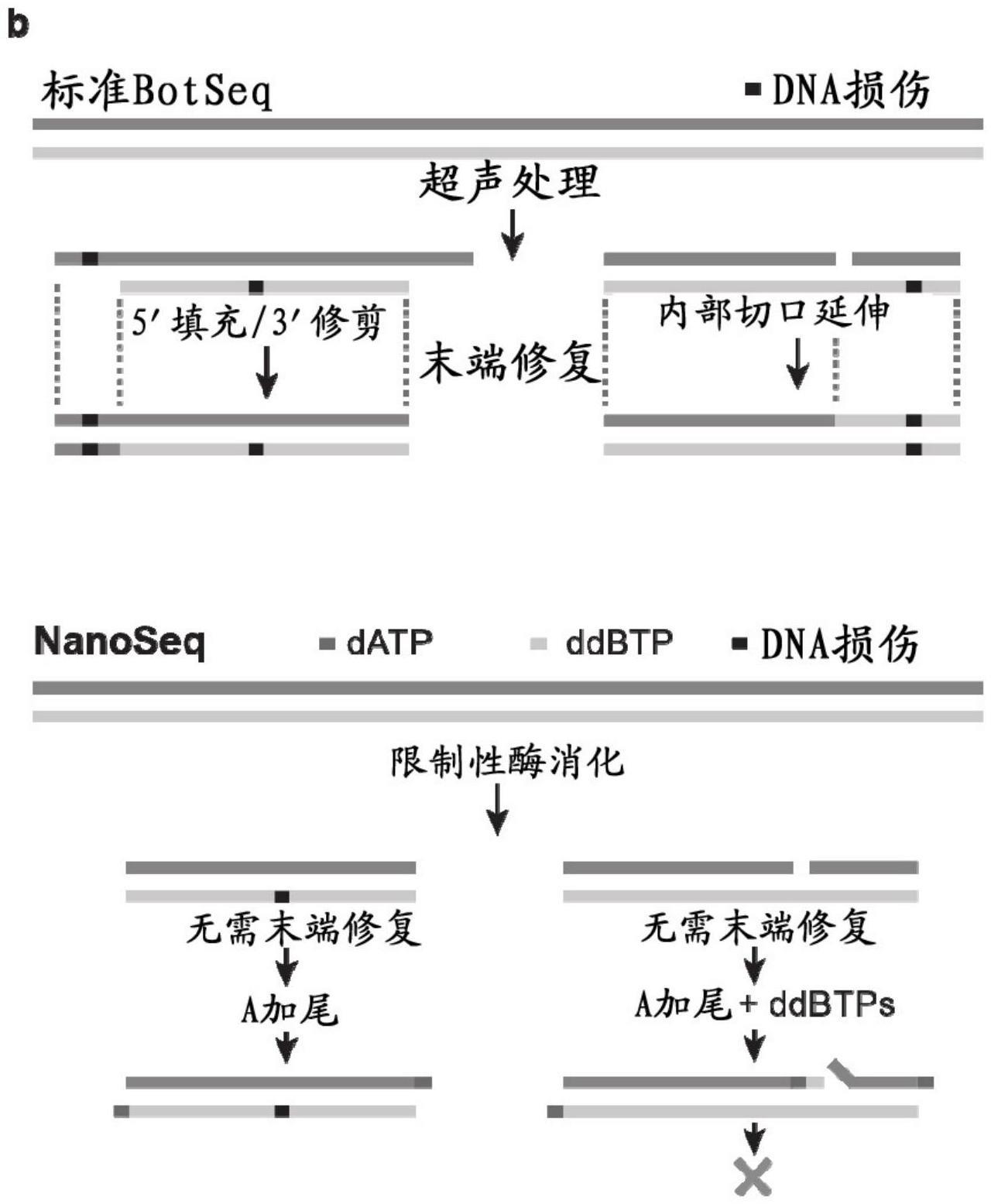
2、用于研究遗传异源样品的标准测序方法的基本限制是由于这些技术的错误率,需要检测多个细胞中的相同突变以区分真正突变与测序错误。虽然标准测序技术通常的错误率为高于1×10-3个错误/bp,但迄今为止测序的大多数体细胞组织的突变负荷数量级为1×10-8至1×10-6个突变/bp(abascal等人,(2021)doi:https://doi.org/10.1038/s41586-021-03477-4;moore等人,(2021)doi:https://doi.org/10.1038/s41586-021-03822-7;以及cagan等人,(2021)doi:https://doi.org/10.1101/2021.08.19.456982)。已经开发了几种方案,通过用唯一的分子条形码标记dna的单个分子并多次读取相同的分子来提高标准测序方法的准确性,通过单分子共有序列测序来降低错误率(hiatt等人,(2010)doi:https://doi.org/10.1038/nmeth.1416,casbon等人,(2011)doi:https://doi.org/10.1093/nar/gkr217,kinde等人,(2011)doi:https://doi.org/10.1073/pnas.1105422108)。最准确的方法使用双链共有测序(schmitt等人,(2012)doi:https://doi.org/10.1073/pnas.1208715109),对dna分子的两条链的拷贝进行测序以去除存在于单个读段中的测序错误和存在于两条链之一的拷贝中的pcr错误。然而,作图错误——特别是在基因组的重复区域中——和在文库制备期间链之间的错误的意外复制违反了两条链的独立性并限制了这些方法的准确性(abascal等人,(2021))。据报道,使用超声处理和末端修复的标准双链测序的错误率高于10-7个错误/bp(abascal等人,(2021))。
3、因此,需要具有更高准确性的方法,例如生成具有错误小于10-7/bp的序列数据的方法,以便能够准确检测和鉴定来自细胞群体的体细胞突变。
技术实现思路
1、根据本发明的第一方面,提供了一种生成用于测序的核酸文库的方法,该方法包括以下步骤:
2、(i)将核酸组合物片段化以生成具有平末端的片段化核酸分子;
3、(ii)在存在于具有平末端的片段化核酸分子中的内部切口位点处引入双脱氧核苷酸并加尾以生成加尾的核酸片段;以及
4、(iii)将测序接头添加至加尾的核酸片段以生成核酸文库。
5、在一些实施方案中,步骤(i)中将核酸组合物片段化包括使用一种或多种生成平末端/非粘性末端的核酸内切酶。在其他实施方案中,将核酸组合物片段化包括机械片段化(例如,超声处理),并使用一种或多种核酸外切酶(例如,绿豆核酸酶)从片段化核酸分子中去除突出端(overhangs),以生成具有平末端的片段化核酸分子。
6、因此,根据本发明的另一个方面,提供了一种用于生成用于测序的核酸文库的方法,该方法包括以下步骤:
7、(ia)通过超声处理使核酸组合物片段化以生成片段化核酸分子;
8、(ib)使用一种或多种核酸外切酶去除片段化核酸分子的突出端以生成具有平末端的片段化核酸分子;
9、(ii)在存在于片段化核酸分子中的内部切口位点处引入双脱氧核苷酸并加尾以生成加尾的核酸片段;以及
10、(iii)将测序接头添加至加尾的核酸片段以生成核酸文库。
11、在又一实施方案中,该方法还包括以下步骤:
12、(iv)选择性地富集对应于一个或多个目的基因组区域的核酸以生成富集的核酸文库。
13、根据本发明的另一方面,提供了一种用于检测单个dna分子中存在的突变的方法,该方法包括以下步骤:
14、(i)使用本文所述的方法生成核酸文库或富集的核酸文库;
15、(ii)对核酸文库或富集的核酸文库进行测序以生成测序数据;以及
16、(iii)计算分析步骤(ii)的测序数据以检测单个dna分子中突变的存在和/或特征(identity)。
17、在另一方面,提供了一种用于检测存在于来自细胞群体的单个dna分子中的突变的方法,包括进行本文描述的方法。
18、在又一方面,提供了一种诊断人类或动物受试者中疾病的方法,所述方法包括以下步骤:
19、(i)根据本文所述的方法检测单个dna分子中突变的存在和/或特征;以及
20、(ii)使用突变的存在和/或特征作为人类或动物受试者中疾病的指标。
21、在另一方面,提供了一种用于检测细胞中可疑突变的存在和/或特征的体外方法,该方法包括以下步骤:
22、(i)在体外培养细胞和/或使体外细胞培养物经受诱变过程,例如通过进行基因编辑;以及
23、(ii)根据本文所述的方法,检测细胞或细胞培养物中单个dna分子中突变的存在和/或特征。
- 还没有人留言评论。精彩留言会获得点赞!