可发泡PMI前驱体珠粒及基于水相悬浮法和超临界CO2的制备方法
可发泡pmi前驱体珠粒及基于水相悬浮法和超临界co2的制备方法
技术领域
1.本发明属于高分子发泡材料制备领域,涉及一种可发泡pmi前驱体珠粒及基于水相悬浮法和超临界co2的制备方法。
背景技术:
2.聚甲基丙烯酰亚胺(polymerthacrylimide,pmi)泡沫是当前高性能夹层结构复合材料泡沫芯材的最主要品种。pmi泡沫以(甲基)丙烯酸(maa/aa)和(甲基)丙烯腈(man/an)为主单体,以(甲基)丙烯酰胺(mam/am)和(甲基)丙烯酸酯(mma/ma)类为辅助单体,以自由基引发剂、物理发泡剂(甲酰胺、低分子醇)和其它助剂作为原料,将原料混合液在封闭平板模具中于35~60℃水浴本体聚合成为可发泡共聚物板材,再经180~220℃高温发泡和后处理而成为pmi泡沫板(张广成,等.橡塑技术与装备.2021,47(10):23-30)。高温过程发泡和后处理过程中,利用大分子链内相邻-cn与-cooh重排酰亚胺化、相邻-cooh与-conh2脱水酰亚胺化、相邻-coor与-conh2脱醇酰亚胺化等化学反应实现大分子内环化和大分子间的交联,从而得到耐热性能和力学性能十分优异的pmi泡沫板材,并作为夹层结构复合材料的芯材广泛应用于航空航天、交通运输、风电叶片、医疗器械、体育器材等高新技术领域。
3.但是,上述pmi泡沫制备技术却存在如下主要缺点:第一,自由基本体聚合过程中反应热难以排除,在大厚度、大面积制品中容易产生爆聚;第二,封闭静止聚合过程造成补加单体困难,大分子链的序列结构可调控性差;第三,原料甲酰胺类发泡剂毒性大;第四,聚合时间长达几天到十几天,生产效率低,产品成本很高;第五,只能得到板材类产品,不能直接制备形状复杂的零部件。
4.为了满足制备形状复杂的pmi泡沫制品的需求,目前通用的方法是将可发泡pmi聚合板材粉碎,进而放入模内进行高温发泡得到相应的pmi泡沫制品。
5.文献1(中国专利,cn101857656b)将aa或maa、an或man、引发剂、分子量调节剂、发泡剂、泡沫稳定剂和交联剂置于平板模具,先在30~50℃低温本体预聚10~72h,再在50~100℃高温聚合1~30h,将聚合板材粉碎后得到可膨胀颗粒,然后放入模具升温到100~180℃预发泡0.1~2h、190~260℃发泡0.1~10h、并在150~230℃熟化1~5h制备模内pmi泡沫。这一方法过程复杂、环保性差、颗粒尺寸难以控制、颗粒之间粘结性差、发泡倍率低,所得模内pmi泡沫的力学性能很差,无法实际使用。
6.文献2(中国专利,cn103232568b.)将an或man与maa、引发剂、交联剂混合后通过紫外光辐射引发聚合,当聚合转化率达到60%~80%时进行固液分离,再将固体通过减压蒸馏除去未反应的组分得到固体共聚物,固体共聚物在液氮中粉碎后再吸附叔丁醇,然后热压发泡、后处理得到pmi泡沫板。这一方法同样存在过程复杂、环保性差、颗粒尺寸难以控制、固体颗粒吸附发泡剂能力低、颗粒之间粘结性差等问题,所得热压pmi泡沫的力学性能和耐热性能不高,无法实际使用。
7.文献3(中国专利,cn106349419b)将mam和maa、引发剂、分子量调节剂和溶剂混合
后共聚,然后向共聚物中加入脱水剂进行酰胺基脱水反应和减压蒸馏得到共聚物,共聚物粉碎后与发泡剂混合进行预发泡,预发泡板破碎成为1~5mm后,加入模具进一步热发泡成为模内pmi泡沫制品。这一方法虽然避免使用毒性较大的丙烯腈类单体,但制备过程复杂而且分子结构中环化率与交联度很低,泡沫的力学性能和耐热性能很低,无法实际使用。
8.文献4(中国专利,cn103421206b.)报道了一种基于水相悬浮聚合制备an/maa共聚物,然后将共聚物干燥粉碎后再吸附碳酰胺发泡剂,模压发泡制备pmi泡沫。这一方法虽然有利于克服传统pmi前驱体预聚过程中容易出现爆聚的缺点,但所得共聚物粉末吸附碳酰胺的能力极小,所得模压pmi泡沫发泡倍率、力学性能和耐热性均很低,无法实际使用。
9.文献5(中国专利,cn108409999a.)通过水相悬浮聚合制备pmi前驱体,再在前驱体中加入交联剂、发泡剂通过轧辊去除水分后得到薄片状混合物,进一步模压发泡得到pmi泡沫。这一方法发泡倍率极低,泡沫结构和性能很难被有效控制,只能得到密度250kg/m3以上的模内pmi泡沫,无法实际使用。
10.文献6(中国专利,cn104995243a)在60~100℃水相悬浮聚合制备0.2~0.6mm可膨胀pmi前驱体珠粒,其中油相含有主单体maa或aa、man或an、可共聚单体甲基丙烯酸叔丁酯、引发剂偶氮二异丁腈(aibn)以及物理发泡剂c3~c7醇等,水相为分散剂硫酸钠、聚甲基丙烯酸等。可膨胀前驱体珠粒在模具内经过150~250℃高温发泡得到100~300kg/m3的模内pmi泡沫制品。这一方法将甲基丙烯酸叔丁酯引入到聚合物主链中会降低聚合物基体的力学性能以及耐热性,同时物理发泡剂c3~c7醇与聚合物基体相容性差,在基体中分布不均,会在聚合物基体中引起缺陷,且珠粒发泡后孔径不均一,极易出现很大的泡孔,这对泡沫材料的力学性能极为不利。该方法无法得到密度低于100kg/m3以下的模内pmi泡沫,泡沫的力学性能也低于传统方法,也无法实际使用。
11.上述报道制备可发泡pmi前驱体珠粒的方法,多数是将可发泡pmi前驱体板材进行粉碎进而发泡制备模内发泡产品,这类方法所得颗粒尺寸极不规则,发泡能力低,泡沫块材力学性能很差,环境污染大。进一步采用悬浮聚合方法虽然可以得到可发泡pmi前驱体珠粒,但是所用发泡剂甲基丙烯酸叔丁酯和醇类的引入对发泡块材的力学性能下降影响很大,泡沫的膨胀倍率也较低。
技术实现要素:
12.要解决的技术问题
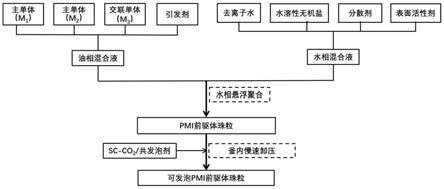
13.为了避免现有技术的不足之处,本发明提出一种可发泡pmi前驱体珠粒及基于水相悬浮法和超临界co2的制备方法,用于解决现有pmi前驱体聚合板粉碎方法制备模内pmi泡沫制品存在的过程复杂、不环保、发泡倍率低、颗粒尺寸难以控制、颗粒之间粘结性差、泡沫性能低等技术难题。也用于解决现有水相悬浮法制备可发泡pmi前驱体珠粒采用甲基丙烯酸叔丁酯和醇类发泡剂带来的泡沫力学性能和耐热性低、发泡倍率不高的难题。本发明提供一种基于水相悬浮法和超临界co2制备可发泡pmi前驱体珠粒的方法,技术方案是采用水相悬浮聚合先制备不含发泡剂的pmi前驱体珠粒,再经过高压釜进行气体饱和制备可发泡pmi前驱体珠粒。
14.技术方案
15.一种可发泡pmi前驱体珠粒,其特征在于包括油相和水相的混合物,其中油相占水
相的质量比为10%~35%;所述油相中各组分的重量比:40~65%的主单体m1、30~60%的主单体m2、0~10%的交联剂和0.01~0.5%的引发剂;所述水相中各组分重量比占去离子水的比例如下:10~40%的水溶性无机盐、0.05~3%的分散剂、0.02~1%的表面活性剂;所述主单体m1为甲基丙烯酸或者丙烯酸;所述主单体m2为甲基丙烯腈或者丙烯腈;所述可发泡pmi前驱体珠粒直径为0.8~12mm,在160~240℃的温度下进行模内发泡。
16.所述交联剂为(甲基)丙烯酰胺、丙烯酸镁、甲基丙烯酸镁、丙烯酸锌、甲基丙烯酸锌或双马来酰亚胺。
17.所述引发剂为偶氮二异丁腈aibn、偶氮二异庚腈aivn、过氧化二苯甲酰bpo、过氧化十二酰lpo、过氧化甲乙酮mekp、过氧化叔戊酸叔丁酯tbpv、过氧化二碳酸二异丙酯ipp或过氧化二碳酸二环己酯dcpd。
18.所述水溶性无机盐为氯化钠、硫酸钠、硝酸钠、氯化钾、硫酸钾或硝酸钾。
19.所述分散剂为聚乙烯醇、丙基纤维素、羟丙基甲基纤维素、聚(甲基)丙烯酸、明胶、甲基纤维素、羧甲基纤维素、聚乙烯吡咯烷酮、乙基纤维素、羟乙基纤维素、聚乙二醇、甲基纤维素或羧甲基纤维素。
20.所述表面活性剂为十二烷基苯磺酸钠、十二烷基硫酸钠、硬脂酸、脂肪酸甘油酯或脂肪酸山梨坦。
21.一种基于水相悬浮法和超临界co2制备所述可发泡pmi前驱体珠粒的方法,其特征在于步骤如下:
22.步骤1:将主单体m1、主单体m2、交联剂和引发剂,按照重量比:40~70%的主单体m1、30~60%的主单体m2、0~10%的交联剂和0.01~0.5%的引发剂混合均匀,配制成油相;
23.步骤2:将去离子水、水溶性无机盐、分散剂、表面活性剂,按照10~40%的水溶性无机盐、0.05~3%的分散剂、0.02~1%的表面活性剂和余量为去离子水混合均匀,配制成水相;
24.步骤3:将水相倒入反应釜中搅拌,控制反应釜温度在25~45℃,向反应釜内加入油相,继续搅拌5~60min提高反应温度至45~75℃,保持反应3~20h,随后升高反应温度至75~95℃进行熟化处理1h后自然冷却至室温,得聚合物珠粒;
25.所述油相占水相的质量比为10%~35%;
26.步骤4:将反应后的聚合物珠粒通过金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;所述pmi前驱体珠粒直径为0.5~10mm;
27.步骤5:将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入sc-co2的共发泡剂,并保持高压釜内压力8~30mpa,温度35~135℃,饱和时间3~30h;
28.所述sc-co2的共发泡剂的含量能够浸没整个模具;
29.随后对高压釜进行卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到发泡pmi前驱体珠粒;所述可发泡pmi前驱体珠粒直径为0.8~12mm。
30.所述搅拌速度100~500rpm。
31.所述筛网为500μm。
32.所述sc-co2的共发泡剂包括戊烷、乙醇、水、异辛烷、甲醇、正丙醇、异丙醇、正丁醇或异丁醇。
33.有益效果
34.本发明提出的一种可发泡pmi前驱体珠粒及基于水相悬浮法和超临界co2的制备方法,将主单体m1(甲基丙烯酸)或者丙烯酸)、主单体m2(甲基丙烯腈或者丙烯腈)、交联单剂、引发剂配制成油相;将去离子水、水溶性无机盐、分散剂、表面活性剂,配制成水相;将水相与油相反应后自然冷却至室温;将反应后的pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入共发泡剂,实现pmi前驱体珠粒的饱和吸附。随后对高压釜进行慢速卸压发泡,完成后取出釜内珠粒,从而得到后续可以在模内进行热发泡的可发泡pmi前驱体珠粒。
35.与传统的本体聚合和悬浮聚合制备可发泡pmi前驱体相比,本发明的技术优势是:第一,通过引入适量水溶性无机盐解决了水溶性单体如(甲基)丙烯酸、(甲基)丙烯酰胺在水相中的溶解难题,避免了油相中缺少这类单体所造成的共聚物中分子链环化率和交联度低所造成的泡沫力学性能低和耐热性下降的缺点。第二,通过选用合适的表面活性剂、分散剂、组份含量控制以及水相悬浮工艺参数控制可以得到粒径可控的球形珠粒,有利于后续模内发泡时珠粒界面之间的粘结,有利于制备形状复杂的pmi泡沫制品。第三,本发明没有使用对耐热性和力学性能不利的甲基丙烯酸叔丁酯类单体,珠粒中可发性气体主要来自于环境友好的超临界co2,避免了使用传统甲酰胺带来的毒性问题,辅助物理发泡剂戊烷、乙醇、水、异辛烷、甲醇、正丙醇、异丙醇、正丁醇、异丁醇等提供了更加高的发泡倍率,能够得到密度低于100kg/m3的模内pmi泡沫制品。第四,本发明采用水相悬浮聚合先制备不含发泡剂的pmi前驱体珠粒,再经过高压釜进行气体饱和制备可发泡pmi前驱体珠粒,这种两步法工艺不仅容易控制前驱体珠粒的分子结构以及球形颗粒的尺寸和分布,而且不含有毒性大的甲酰胺类发泡剂,具有环保性好、有利于后续工业化生产。
36.本发明所述可发泡pmi前驱体珠粒的直径为0.8~12mm,可以在160~240℃的温度下进行模内发泡,进而制备pmi模内泡沫制品,克服目前pmi泡沫全部是泡沫板的单一现状,从而有望应用于汽车工业、航空航天、医疗器械、轨道交通、风力发电等领域对复杂pmi泡沫产品的需求。
附图说明
37.图1为实施例1所制备的pmi前驱体珠粒、可发泡pmi前驱体珠粒以及经过高温模内发泡后所得pmi泡沫的照片;
38.a、pmi前驱体珠粒(平均粒径2.6mm);b、可发泡pmi前驱体珠粒(平均粒径3.0mm);c、模内发泡得到的样品;
39.图2为实施例2所制备的pmi前驱体珠粒、可发泡pmi前驱体珠粒以及经过高温模内发泡后所得pmi泡沫的照片;
40.a、pmi前驱体珠粒(平均粒径2.4mm);b、可发泡pmi前驱体珠粒(平均粒径2.8mm);c、模内发泡得到的样品;
41.图3为实施例3所制备的pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片;
42.a、pmi前驱体珠粒(平均粒径3.1mm);b、可发泡pmi前驱体珠粒(平均粒径4.3mm);
43.图4为实施例4所制备的pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片;
44.a、pmi前驱体珠粒(平均粒径4.5mm);b、可发泡pmi前驱体珠粒(平均粒径5.0mm);
45.图5为实施例5所制备的pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片;
46.a、pmi前驱体珠粒(平均粒径1.6mm);b、可发泡pmi前驱体珠粒(平均粒径1.9mm);
47.图6为实施例6所制备的pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片;
48.a、pmi前驱体珠粒(平均粒径1.0mm);b、可发泡pmi前驱体珠粒(平均粒径1.2mm);
49.图7为对比例1经水相悬浮聚合所制备的团聚体的照片。
50.图8为制备方法流程图
具体实施方式
51.现结合实施例、附图对本发明作进一步描述:
52.以下将对本发明提供的一种基于水相悬浮法和超临界co2制备可发泡pmi前驱体珠粒的方法作进一步说明。
53.本发明提供的一种基于水相悬浮法和超临界co2制备可发泡pmi前驱体珠粒的方法包括以下步骤:
54.(a)将主单体m1(甲基丙烯酸或者丙烯酸)、主单体m2(甲基丙烯腈或者丙烯腈)、交联单体m3、引发剂,按照计量比混合均匀,配制成油相;
55.(b)将去离子水、水溶性无机盐、分散剂、表面活性剂,按照计量比混合均匀,配制成水相;
56.(c)将水相倒入反应釜中,控制搅拌速度100~500rpm,控制反应釜温度在25~45℃,向反应釜内加入油相,继续搅拌5~60min提高反应温度至45~75℃,保持反应3~20h,随后升高反应温度至75~95℃进行熟化处理1h后自然冷却至室温;
57.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
58.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷、乙醇、水、异辛烷、甲醇、正丙醇、异丙醇、正丁醇、异丁醇中的至少一种作为sc-co2的共发泡剂。并保持高压釜内压力8~30mpa,温度35~135℃,饱和时间3~30h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
59.其中,步骤(a)中所述交联单体m3为(甲基)丙烯酰胺、丙烯酸镁、甲基丙烯酸镁、丙烯酸锌、甲基丙烯酸锌、双马来酰亚胺中的至少一种,所述交联剂可以具体细分为两类:第一类单体为丙烯酸镁、甲基丙烯酸镁、丙烯酸锌、甲基丙烯酸锌、双马来酰亚胺等,这种单体两端含有双键,在珠粒聚合过程中即形成交联结构。第二类为(甲基)丙烯酰胺这一类对应每个单体仅含有一个双键,在珠粒合成后并没有形成双键,仅在在珠粒高温发泡过程中大分子链之间的酰胺基相互反应生成交联结构进而支撑泡孔结构,可以避免发泡过程中出现泡孔塌缩的问题。
60.步骤(a)中所述引发剂为偶氮二异丁腈(aibn)、偶氮二异庚腈(aivn)、过氧化二苯甲酰(bpo)、过氧化十二酰(lpo)、过氧化甲乙酮(mekp)、过氧化叔戊酸叔丁酯(tbpv)、过氧化二碳酸二异丙酯(ipp)、过氧化二碳酸二环己酯(dcpd)中的至少一种,引发剂的半衰期温度要和聚合温度相匹配,以使聚合反应不至于太快,避免分子量过低。而多种引发剂的协同使用对于聚合单体的完全反应比较有利,可以减少单体残留,提高共聚物分子量,制备出高耐热、高力学性能的pmi前驱体珠粒。
61.步骤(a)中所述油相中各组分的配比按照重量比如下
62.40~65%(甲基)丙烯酸;
63.30~60%(甲基)丙烯腈;
64.0~10%交联剂;
65.和0.01~0.5%引发剂。
66.主单体(甲基)丙烯酸和(甲基)丙烯腈的比例配比要合适,以便使共聚物化学主链结构中形成较多的(甲基)丙烯酸和(甲基)丙烯腈交替共聚的单元,从而在后续高温发泡的过程中在主链上形成更多的酰亚胺环,提高材料的耐热性和力学性能。交联剂的含量也应该合理,过多的交联剂不利于发泡倍率提高,过低的交联剂对泡沫制品的力学性能和耐热性不利。
67.步骤(b)中所述水溶性无机盐为氯化钠、硫酸钠、硝酸钠、氯化钾、硫酸钾、硝酸钾中的至少一种,主要起到降低水溶性单体在水中溶解度的问题。
68.步骤(b)中所述分散剂为聚乙烯醇、丙基纤维素、羟丙基甲基纤维素、聚(甲基)丙烯酸、明胶、甲基纤维素、羧甲基纤维素、聚乙烯吡咯烷酮、乙基纤维素、羟乙基纤维素、聚乙二醇、甲基纤维素、羧甲基纤维素中的至少一种。分散剂吸附在被机械搅拌形成的液滴表面,形成溶剂化层,起保护油相悬浮液滴的稳定作用。分散剂的合理选用对于制备出形状规则,表面光滑,粒径均匀的pmi珠粒至关重要。
69.步骤(b)中所述表面活性剂为十二烷基苯磺酸钠、十二烷基硫酸钠、硬脂酸、脂肪酸甘油酯、脂肪酸山梨坦中的至少一种。这一类表面活性剂分子中同时具有亲水基和亲油基,它聚集在油/水界面上,可以降低界面张力和减少形成乳状液所需要的能量,从而提高乳状液的能力。
70.步骤(b)中所述水相中各组分按照重量比占去离子水的比例如下
71.10~40%水溶性无机盐;
72.0.05~3%分散剂;
73.和0.02~1%表面活性剂。
74.步骤(d)中所述pmi前驱体珠粒直径为0.5~10mm,烘干温度为60~100℃。步骤(e)中所述可发泡pmi前驱体珠粒直径为0.8~12mm,可以在160~240℃的温度下进行模内发泡,进而制备pmi模内泡沫制品。
75.以下将通过具体实施例对所述一种基于水相悬浮法和超临界co2制备可发泡pmi前驱体珠粒的方法做进一步的说明。
76.实施例1:
77.(a)将主单体甲基丙烯酸20ml、丙烯腈16ml,交联单体甲基丙烯酰胺0.35g,引发剂偶氮二异丁腈0.1g,按照计量比配成单体混合液,配制成油相;
78.(b)将去离子水200ml、水溶性无机盐氯化钠50g,分散剂羟乙基纤维素2g,表面活性剂十二烷基苯磺酸钠1g,按照计量,混合均匀,配制水相;
79.(c)反应过程将水相倒入反应釜中,控制搅拌速度320rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
80.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
81.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间18h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
82.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图1中左右两图所示,pmi前驱体珠粒的尺寸为2.6mm,可发泡pmi前驱体珠粒的尺寸为3.0mm。将所得可发泡pmi前驱体放进模具中于200℃进行高温处理30min后得到密度0.15g/cm3的pmi泡沫如图1所示
83.实施例2:
84.(a)将主单体甲基丙烯酸20ml、丙烯腈16ml,交联单体甲基丙烯酰胺0.35g,引发剂偶氮二异丁腈0.1g,按照计量比配成单体混合液,配制成油相;
85.(b)将去离子水200ml、水溶性无机盐氯化钠50g,分散剂羟丙基甲基纤维素2g,表面活性剂十二烷基苯磺酸钠1g,按照计量,混合均匀,配制水相;
86.(c)反应过程将水相倒入反应釜中,控制搅拌速度280rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至65℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
87.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
88.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力15mpa,温度80℃,饱和时间19h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
89.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图2中左右两图所示,pmi前驱体珠粒的尺寸为2.4mm,可发泡pmi前驱体珠粒的尺寸为2.8mm。将所得可发泡pmi前驱体放进模具中于200℃进行高温处理30min后得到密度0.10g/cm3的pmi泡沫如图2所示
90.实施例3:
91.(a)将主单体甲基丙烯酸22ml、丙烯腈19ml,交联单体丙烯酰胺0.65g,引发剂偶氮二异庚腈0.15g,按照计量比配成单体混合液,配制成油相;
92.(b)将去离子水170ml、水溶性无机盐氯化钠60g,分散剂聚乙烯醇3g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
93.(c)反应过程将水相倒入反应釜中,控制搅拌速度180rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至70℃,保持反应20h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
94.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
95.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度120℃,饱和时间16h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
96.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图3中左右两图所示,pmi前驱体珠粒的尺寸为3.1mm,可发泡pmi前驱体珠粒的尺寸为4.3mm。
97.实施例4:
98.(a)将主单体甲基丙烯酸20ml、丙烯腈16ml,交联单体甲基丙烯酰胺0.35g,引发剂偶氮二异丁腈0.1g,按照计量比配成单体混合液,配制成油相;
99.(b)将去离子水150ml、水溶性无机盐氯化钠50g,分散剂聚乙烯醇2g,表面活性剂十二烷基苯磺酸钠1g,按照计量,混合均匀,配制水相;
100.(c)反应过程将水相倒入反应釜中,控制搅拌速度160rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至65℃,保持反应10h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
101.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
102.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度100℃,饱和时间22h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
103.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图4中左右两图所示,pmi前驱体珠粒的尺寸为4.5mm,可发泡pmi前驱体珠粒的尺寸为5.0mm。
104.实施例5:
105.(a)将主单体甲基丙烯酸20ml、丙烯腈20ml,交联单体甲基丙烯酰胺0.35g,引发剂偶氮二异丁腈0.06g,按照计量比配成单体混合液,配制成油相;
106.(b)将去离子水200ml、水溶性无机盐氯化钠40g,分散剂聚乙烯吡咯烷酮2g,表面活性剂十二烷基硫酸钠0.5g,按照计量,混合均匀,配制水相;
107.(c)反应过程将水相倒入反应釜中,控制搅拌速度400rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌30min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
108.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
109.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力15mpa,温度80℃,饱和时间24h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
110.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图5中左右两图所示,pmi前驱体珠粒的尺寸为1.6mm,可发泡pmi前驱体珠粒的尺寸为1.9mm。
111.实施例6:
112.(a)将主单体甲基丙烯酸18ml、丙烯腈20ml,交联单体甲基丙烯酰胺1.2g,引发剂偶氮二异丁腈0.08g,按照计量比配成单体混合液,配制成油相;
113.(b)将去离子水200ml、水溶性无机盐氯化钠45g,分散剂甲基纤维素2g,表面活性剂十二烷基苯磺酸钠1g,按照计量,混合均匀,配制水相;
114.(c)反应过程将水相倒入反应釜中,控制搅拌速度450rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应15h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
115.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从
而得到pmi前驱体珠粒;
116.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间20h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
117.所得pmi前驱体珠粒的照片和可发泡pmi前驱体珠粒的照片分别如图6中左右两图所示,pmi前驱体珠粒的尺寸为1.0mm,可发泡pmi前驱体珠粒的尺寸为1.2mm。
118.实施例7:
119.(a)将主单体甲基丙烯酸17ml、丙烯腈15ml,交联单体甲基丙烯酸锌1.4g,引发剂过氧化二碳酸二异丙酯0.16g,按照计量比配成单体混合液,配制成油相;
120.(b)将去离子水160ml、水溶性无机盐氯化钠60g,分散剂聚甲基丙烯酸1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
121.(c)反应过程将水相倒入反应釜中,控制搅拌速度220rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至66℃,保持反应18h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
122.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
123.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
124.所得pmi前驱体珠粒的尺寸为2.7mm,可发泡pmi前驱体珠粒的尺寸为3.1mm
125.实施例8:
126.(a)将主单体甲基丙烯酸23ml、丙烯腈21ml,交联单体甲基丙烯酸镁1.4g,引发剂偶氮二异丁腈0.16g,按照计量比配成单体混合液,配制成油相;
127.(b)将去离子水200ml、水溶性无机盐氯化钠60g,分散剂甲基纤维素1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
128.(c)反应过程将水相倒入反应釜中,控制搅拌速度440rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
129.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
130.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
131.所得pmi前驱体珠粒的尺寸为1.3mm,可发泡pmi前驱体珠粒的尺寸为1.6mm
132.实施例9:
133.(a)将主单体甲基丙烯酸17ml、丙烯腈18ml,交联单体甲基丙烯酰胺2.4g,引发剂偶氮二异丁腈0.15g,按照计量比配成单体混合液,配制成油相;
134.(b)将去离子水200ml、水溶性无机盐氯化钠60g,分散剂羟丙基甲基纤维素1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
135.(c)反应过程将水相倒入反应釜中,控制搅拌速度320rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至70℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
136.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
137.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
138.所得pmi前驱体珠粒的尺寸为2.0mm,可发泡pmi前驱体珠粒的尺寸为2.1mm
139.实施例10:
140.(a)将主单体甲基丙烯酸17ml、丙烯腈19ml,交联单体丙烯酸锌1.0g,引发剂偶氮二异丁腈0.16g,按照计量比配成单体混合液,配制成油相;
141.(b)将去离子水200ml、水溶性无机盐氯化钠55g,分散剂羟丙基甲基纤维素1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
142.(c)反应过程将水相倒入反应釜中,控制搅拌速度220rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
143.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
144.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力25mpa,温度80℃,饱和时间18h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
145.所得pmi前驱体珠粒的尺寸为2.8mm,可发泡pmi前驱体珠粒的尺寸为3.2mm
146.实施例11:
147.(a)将主单体甲基丙烯酸19ml、丙烯腈19ml,交联单体甲基丙烯酰胺1.4g,引发剂偶氮二异丁腈0.16g,按照计量比配成单体混合液,配制成油相;
148.(b)将去离子水200ml、水溶性无机盐氯化钠60g,分散剂羟乙基纤维素1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
149.(c)反应过程将水相倒入反应釜中,控制搅拌速度290rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至72℃,保持反应8h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
150.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
151.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
152.所得pmi前驱体珠粒的尺寸为2.3mm,可发泡pmi前驱体珠粒的尺寸为2.5mm
153.实施例12:
154.(a)将主单体甲基丙烯酸20ml、丙烯腈15ml,交联单体甲基丙烯酰胺1.4g,引发剂
偶氮二异丁腈0.08g,按照计量比配成单体混合液,配制成油相;
155.(b)将去离子水200ml、水溶性无机盐氯化钠35g,分散剂羟乙基纤维素1g,表面活性剂十二烷基苯磺酸钠0.2g,按照计量,混合均匀,配制水相;
156.(c)反应过程将水相倒入反应釜中,控制搅拌速度160rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
157.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
158.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
159.所得pmi前驱体珠粒的尺寸为4.7mm,可发泡pmi前驱体珠粒的尺寸为5.1mm
160.对比例1:
161.(a)将主单体甲基丙烯酸17ml、丙烯腈15ml,交联单体甲基丙烯酰胺0.64g,引发剂偶氮二异丁腈0.06g,按照计量比配成单体混合液,配制成油相;
162.(b)将去离子水200ml、水溶性无机盐氯化钠10g,分散剂羟乙基纤维素1g,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
163.(c)反应过程将水相倒入反应釜中,控制搅拌速度220rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
164.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
165.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
166.由于水溶性无机盐硫酸钠加入量过少,并未出现pmi前驱体珠粒,反应釜内出现团聚体,团聚体的如图7所示。
167.对比例2:
168.(a)将主单体甲基丙烯酸17ml、丙烯腈15ml,交联单体甲基丙烯酰胺0.64g,引发剂偶氮二异丁腈0.06g,按照计量比配成单体混合液,配制成油相;
169.(b)将去离子水200ml、水溶性无机盐氯化钠60g,分散剂不加,表面活性剂十二烷基苯磺酸钠0.5g,按照计量,混合均匀,配制水相;
170.(c)反应过程将水相倒入反应釜中,控制搅拌速度220rpm,控制反应釜温度在30℃,向反应釜内加入油相,继续搅拌20min提高反应温度至60℃,保持反应5h,随后升高反应温度至80℃进行熟化处理1h后自然冷却至室温;
171.(d)将反应后的聚合物珠粒通过500μm金属筛网过滤,舍弃结块材料,清洗,烘干从而得到pmi前驱体珠粒;
172.(e)将pmi前驱体珠粒置于超临界co2高压釜内,向釜内加入一定量的戊烷。并保持高压釜内压力20mpa,温度80℃,饱和时间10h。随后对高压釜进行慢速卸压发泡,卸压速度
小于1mpa/min,卸压完成后取出釜内珠粒,从而得到可发泡pmi前驱体珠粒;
173.由于没有分散剂,并未出现pmi前驱体珠粒,反应釜内出现团聚体。
174.为了进一步说明实施例,此处列出12个实施例与2个对比例的区别表,包括原料和工艺,以及得到的珠粒、发泡珠粒的尺寸等,如表1所示。
175.表1实例汇和对比例对照表
176.177.[0178][0179]
本发明与传统方法中将共聚物板材粉碎成颗粒状进而制备模内pmi泡沫相比,具有显著的优点。
[0180]
一、通过改进水相悬浮聚合工艺和配方,成功制备出球形度较好、尺寸较均一的pmi前驱体珠粒,有利于后续珠粒模内发泡过程中相互之间良好粘结。
[0181]
二、避免使用毒性大的甲酰胺类传统发泡剂,选用的共发泡剂戊烷、乙醇、水等对环境毒性较小且发泡能力强,进一步通过引入超临界co2,提高饱和压力,增强气体在珠粒内的渗透能力。共发泡剂能够与聚合物基体产生相互作用,增大聚合分子链之间的自由体积,有利于促进超临界co2在基体中的溶解度。最终经过慢速卸压后得到的可发泡pmi前驱体珠粒会保留较多的发泡剂(超临界co2和共发泡剂),这对于后续模内升温发泡得到高发泡倍率极为有利,所制备的模内pmi泡沫有望解决现有pmi泡沫产品单一的难题。
[0182]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0183]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1