一种酯型儿茶素-茶氨酸加合物的制备方法
pyrrolidinone-substituted flavan-3-ols with anti-inflammatory activity in lipopolysaccharide-stimulated macrophages are storage-related marker compounds for green tea[j].journal of agricultural andfood chemistry,2020,68(43):12164-12172.)中也鉴定到这类物质,并且作为白茶和绿茶存储的关键标志化合物。
[0004]
2019年,安徽农业大学宛晓春课题组经lc-q-tof-ms、lc-qqq-ms和代谢组学对比研究发现在黄大茶拉老火前后,茶叶中的efsfs类物质含量剧增,推测在黄大茶的高火烘焙中儿茶素与茶氨酸发生了非酶促聚合反应,促使大量epsfs类物质合成(zhou j.,wu y.,long p p.,et al.lc-ms-based metabolomics reveals the chemical changes of polyphenols during high-temperature roasting oflarge-leafyellow tea[j].journal ofagricultural and food chemistry,2019,67(19):5405-5412.)。通过比较等量黄大茶、绿茶、红茶、黑茶提取物对糖尿病模型小鼠糖脂代谢的影响,结果表明,仅黄大茶能够显著降低实验组小鼠的空腹血糖,且小鼠对黄大茶茶汤耐受性更好(10)。同时,饮用黄大茶茶汤还能缓解长期高脂高糖饮食诱导的小鼠非酒精性脂肪肝,降低机体脂质和炎症水平,调控机体糖脂代谢关键基因的表达,说明黄大茶的降血糖效应很可能与大量epsfs类物质合成相关(xu n.,chu j.,dong r.,lu f.,&wan x.yellow tea stimulates thermogenesis in mice through heterogeneous browning of adipose tissues.molecular nutrition&food research,2020:2000864.)。
[0005]
研究人员相继从老白茶、黄大茶中进行epsfs类物质的分离、纯化制备和应用研究,但是天然存在的或者加工过程中形成的epsfs类物质含量很低,且由于茶叶中化合物种类繁多,加工过程复杂,导致对epsfs类物质的分离难度极大,极大限制了该物质在食品和健康领域的应用开发。
技术实现要素:
[0006]
本发明的目的在于提供一种酯型儿茶素-茶氨酸加合物的制备方法,采用体外合成、分离纯化得到酯型儿茶素-茶氨酸加合物,能够推进epsfs类物质在食品和医药领域的开发应用。
[0007]
为了实现上述发明目的,本发明提供以下技术方案:
[0008]
本发明提供了一种酯型儿茶素-茶氨酸加合物的制备方法,包括以下步骤:
[0009]
将表没食子儿茶素没食子酸酯、茶氨酸、ph调节剂和水混合,在密闭和加热条件下进行亲核反应,得到加合产物;
[0010]
将所述加合产物与水混合,进行复溶,将所得加合产物水溶液进行萃取,得到萃取相;所述萃取所用萃取剂为乙酸乙酯或正丁醇;
[0011]
将所述萃取相依次进行旋蒸浓缩和柱层析纯化,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物;
[0012][0013]
优选的,所述表没食子儿茶素没食子酸酯和茶氨酸的质量比为(1~20):(20~1);所述亲核反应过程中,所述水的质量为表没食子儿茶素没食子酸酯和茶氨酸总质量的50~500%。
[0014]
优选的,所述ph调节剂为酸或碱,所述亲核反应的ph值为3~10。
[0015]
优选的,所述加热的温度为60~180℃,所述亲核反应的时间为10~180min。
[0016]
优选的,采用高效液相色谱对所述萃取相进行检测,确定酯型儿茶素-茶氨酸加合物存在;所述高效液相色谱检测所用色谱柱为agilent zorbax sb-aq c18色谱柱,流动相包括a相和b相,所述a相为体积浓度0.2%的甲酸水溶液,b相为甲醇;流速为1ml/min,洗脱条件:
[0017]
0~5min,5%甲酸水溶液~20%甲酸水溶液;
[0018]
5~16min,20%甲酸水溶液~25%甲酸水溶液;
[0019]
16~25min,25%甲酸水溶液;
[0020]
25~38min,25%甲酸水溶液~45%甲酸水溶液;
[0021]
38~40min,45%甲酸水溶液~100%甲醇;
[0022]
40~42min,100%甲醇~5%甲酸水溶液;
[0023]
42~55min,5%甲酸水溶液。
[0024]
优选的,所述柱层析纯化的过程包括:
[0025]
将旋蒸浓缩所得浓缩物溶解于甲醇后,将所得混合液与硅胶混合,蒸干,得到待分离样品;
[0026]
将所述待分离样品进行第一次柱层析分离,得到第一次柱层析分离产物;
[0027]
将所述第一次柱层析分离产物进行薄层色谱分离,将所得分离产物进行第二次柱层析分离,所述第二次柱层析分离包括3个洗脱梯度,得到3个洗脱组分;
[0028]
采用高效液相色谱检测法对所述3个洗脱组分进行检测,确定酯型儿茶素-茶氨酸
加合物所在组分;
[0029]
将酯型儿茶素-茶氨酸加合物所在组分进行高效液相色谱分离,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物。
[0030]
优选的,所述第一次柱层析分离所用填料为硅胶,洗脱试剂为二氯甲烷、甲醇和甲酸,所述二氯甲烷、甲醇和甲酸的体积比为30:10:2。
[0031]
优选的,所述薄层色谱分离所用展开剂为二氯甲烷、甲醇和甲酸的混合溶液,所述混合溶液中甲醇、二氯甲烷和甲酸的体积比为(20~30):(8~10):(0.1~2)。
[0032]
优选的,所述第二次柱层析分离所用填料为ods-c18反向柱;所述第二次柱层析分离包括三个洗脱梯度,洗脱梯度1所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水混合液和甲醇的体积比为95:5;洗脱梯度2所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水溶液和甲醇的体积比为75:25;洗脱梯度3所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水混合液与甲醇的体积比为50:50。
[0033]
优选的,所述高效液相色谱分离所用色谱柱为agilent zorbax sb-aq c18色谱柱,流动相包括a相和b相,所述a相为体积浓度为0.2%的甲酸水溶液,b相为甲醇;所述高效液相色谱分离的洗脱条件为:
[0034]
0~4min,5~25%甲酸水溶液;
[0035]
4~7min,25%~38%甲酸水溶液;
[0036]
7~9min,38%~42%甲酸水溶液;
[0037]
9~17min,42%甲酸水溶液;
[0038]
17~19min,42%甲酸水溶液~100%甲醇;
[0039]
19~20min,100%b相~5%甲酸水溶液;
[0040]
20~25min,5%甲酸水溶液。
[0041]
本发明提供了一种酯型儿茶素-茶氨酸加合物的制备方法,本发明以茶氨酸为底物,将茶氨酸与表没食子儿茶素没食子酸酯(egcg)在高温湿热条件进行亲核反应,egcg中a环6位上的c或8位上的c与茶氨酸的strecker降解产物结合,形成加合产物,将所述加合产物依次通过萃取和分离纯化后得到具有式i、式ii、式iii或式iv所示结构的酯型儿茶素-茶氨酸加合物,即黄烷醇生物碱(n-乙基-2-吡咯烷酮取代的黄烷-3-醇,缩写为epsfs)。本发明通过体外化学合成并分离纯化制备酯型儿茶素-茶氨酸加合物,合成方法和纯化技术操作简单,容易实施,成本较低,且所得酯型儿茶素-茶氨酸加合物具有降糖、降脂等生物活性,能够推进epsfs类物质在食品和医药领域的开发应用。
附图说明
[0042]
图1实施例1中不同反应装置加热后epsfs含量的变化;
[0043]
图2实施例2中不同反应温度对epsfs含量变化的影响;
[0044]
图3实施例3中不同反应时间对epsfs含量变化的影响;
[0045]
图4实施例4中不同ph对epsfs含量变化的影响;
[0046]
图5实施例5中不同加水量对epsfs含量变化的影响;
[0047]
图6实施例6中不同极性试剂对egcg-茶氨酸热反应产物水溶液萃取样色谱图;
[0048]
图7为实施例7中epsfs制备和分离纯化的流程图;
[0049]
图8为实施例7中c18反向柱分离所得组分3中epsfs的色谱图;
[0050]
图9为epsfs1(式i)的质谱离子碎片图;
[0051]
图10为epsfs 2(式ii)的质谱离子碎片图;
[0052]
图11epsfs 3(式iii)的质谱离子碎片图;
[0053]
图12epsfs 4(式iv)的质谱离子碎片图。
具体实施方式
[0054]
本发明提供了一种酯型儿茶素-茶氨酸加合物的制备方法,包括以下步骤:
[0055]
将表没食子儿茶素没食子酸酯、茶氨酸、ph调节剂和水混合,在密闭和加热条件下进行亲核反应,得到加合产物;
[0056]
将所述加合产物与水混合,进行复溶,将所得加合产物水溶液进行萃取,得到萃取相;所述萃取所用萃取剂为乙酸乙酯或正丁醇;
[0057]
将所述萃取相依次进行旋蒸浓缩和柱层析纯化,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物;
[0058][0059][0060]
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
[0061]
本发明将表没食子儿茶素没食子酸酯、茶氨酸、ph调节剂和水混合,在密闭和加热条件下进行亲核反应,得到加合产物。
[0062]
在本发明中,所述表没食子儿茶素没食子酸酯和茶氨酸的纯度均优选为分析纯,纯度优选独立地≥95%。
[0063]
在本发明中,所述表没食子儿茶素没食子酸酯(egcg)和茶氨酸(l-theanine)的质量比优选为(1~20):(20~1),更优选为1:(1~2);所述亲核反应过程中,所述水的质量优选为表没食子儿茶素没食子酸酯和茶氨酸总质量的50~500%,更优选为50~200%。
[0064]
在本发明中,所述ph调节剂优选为酸或碱,所述酸优选为浓度为0.01mol/l的盐酸
溶液,所述碱优选为浓度为0.01mol/l的氢氧化钠溶液,所述亲核反应的ph值优选为3~10。
[0065]
在本发明中,所述表没食子儿茶素没食子酸酯、茶氨酸和水混合的方式优选包括依次进行的涡旋混合和超声混合。在本发明中,所述涡旋混合的速率优选为500r/min,时间优选为2~3min,更优选为2.5min;所述超声混合的功率优选为200w,频率优选为40khz,时间优选为2~3min,更优选为2.5min。本发明通过所述涡旋混合和超声混合,促进原料的溶解。
[0066]
本发明对所述密闭的方式没有特殊的限定,能够实现密闭效果的方式均可;在本发明的实施例中,所述密闭的反应装置为反应釜或密闭玻璃瓶。在本发明中,所述亲核反应的温度优选为60~180℃,更优选为120~150℃;所述亲核反应的时间优选为10~180min,更优选为30~120min,进一步优选为60~90min。在加热的条件下,egcg中a环6位上的c和8位上的c与茶氨酸的strecker降解产物结合,发生亲核反应。
[0067]
得到加合产物后,本发明将所述加合产物与水混合,进行复溶,将所得加合产物水溶液进行萃取,得到萃取相。本发明对所述水的用量没有特殊的限定,能够将加合产物完全复溶即可。本发明对所述加合产物与水混合的过程没有特殊的限定,按照本领域熟知的过程将物料混合均匀即可;在本发明的实施例中,具体是在40℃加热超声混合。
[0068]
在本发明中,所述萃取所用萃取剂优选为乙酸乙酯或正丁醇,所述萃取剂与加合产物水溶液的体积比优选为1:1;所述萃取的次数优选为3次。
[0069]
完成所述萃取后,本发明优选将所得萃取相旋蒸浓缩至干,用甲醇溶解后,采用高效液相色谱检测法进行检测,确定酯型儿茶素-茶氨酸加合物存在;本发明对所述旋蒸浓缩的过程没有特殊的限定,按照本领域熟知的过程进行即可。本发明对所述甲醇的用量没有特殊的限定,能够完全溶剂萃取相即可。
[0070]
在本发明中,所述高效液相色谱检测所用色谱柱优选为agilent zorbax sb-aq c18色谱柱,流动相优选包括a相和b相,所述a相为体积浓度0.2%的甲酸水溶液,b相为甲醇;流速为1ml/min,检测波长为278nm,检测器为dad,柱温为30℃,洗脱条件优选为:
[0071]
0~5min,5%甲酸水溶液~20%甲酸水溶液;
[0072]
5~16min,20%甲酸水溶液~25%甲酸水溶液;
[0073]
16~25min,25%甲酸水溶液;
[0074]
25~38min,25%甲酸水溶液~45%甲酸水溶液;
[0075]
38~40min,45%甲酸水溶液~100%甲醇;
[0076]
40~42min,100%甲醇~5%甲酸水溶液;
[0077]
42~55min,5%甲酸水溶液。
[0078]
本发明优选通过高效液相色谱进行检测,确定酯型儿茶素-茶氨酸加合物存在后,对相应组分进行后续纯化过程。
[0079]
得到萃取相后,本发明将所述萃取相依次进行旋蒸浓缩和柱层析纯化,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物。本发明对所述旋蒸浓缩的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0080]
在本发明中,所述柱层析纯化的过程优选包括:
[0081]
将旋蒸浓缩所得浓缩物溶解于甲醇后,将所得混合液与硅胶混合,蒸干,得到待分离样品;
[0082]
将所述待分离样品进行第一次柱层析分离,得到第一次柱层析分离产物;
[0083]
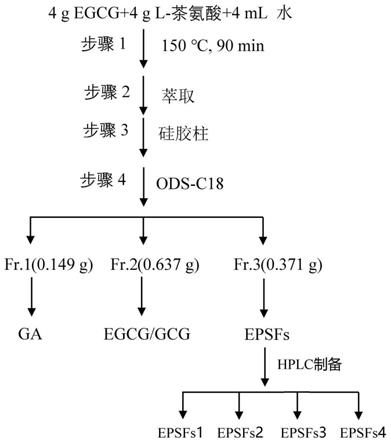
将所述第一次柱层析分离产物进行薄层色谱分离,将所得分离产物进行第二次柱层析分离,所述第二次柱层析分离包括3个洗脱梯度,得到3个洗脱组分;
[0084]
采用高效液相色谱检测法对所述3个洗脱组分进行检测,确定酯型儿茶素-茶氨酸加合物所在组分;
[0085]
将酯型儿茶素-茶氨酸加合物所在组分进行高效液相色谱分离,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物。
[0086]
本发明将旋蒸浓缩所得浓缩物溶解于甲醇后,将所得混合液与硅胶混合,蒸干,得到待分离样品。本发明对所述甲醇的用量没有特殊的限定,能够将浓缩物充分溶解即可,在本发明的实施例中,所述混合液中浓缩物的浓度具体为400mg/ml;本发明优选在水浴锅中将混合液与硅胶的混合物中甲醇挥干,使得混合液中的有效物质被吸附在硅胶粉上。
[0087]
得到待分离样品后,本发明优选将所述待分离样品进行第一次柱层析分离,得到第一次柱层析分离产物。在本发明中,所述第一次柱层析分离的上样方式优选为干法上样;所述第一次柱层析分离所用填料优选为硅胶,洗脱试剂优选为二氯甲烷、甲醇和甲酸,所述二氯甲烷、甲醇和甲酸的体积比优选为30:10:2;玻璃柱规格优选为5
×
40cm。
[0088]
得到第一次柱层析分离产物后,本发明优选将所述得到第一次柱层析分离产物进行薄层色谱分离,将所得分离产物进行第二次柱层析分离,所述第二次柱层析分离包括3个洗脱梯度,得到3个洗脱组分。在本发明中,所述薄层色谱分离所用展开剂优选为二氯甲烷、甲醇和甲酸的混合溶液,所述混合溶液中甲醇、二氯甲烷和甲酸的体积比优选为(20~30):(8~10):(0.1~2),更优选为30:10:2。本发明优选以待分离样品为对照样,对所述第一次柱层析分离产物进行薄层色谱分离,将薄层层析板于254nm的紫外灯下观察斑点分布情况,使用显色剂显色后,将薄层层析板上各流分样品显色后的斑点与对照样的斑点进行对比,将具有相同或者相似斑点的流分进行合并,去除大部分色素。在本发明中,所述显色剂优选为香草醛硫酸乙醇溶液,所述香草醛硫酸乙醇溶液的质量浓度优选为5%。
[0089]
完成所述薄层色谱分离后,本发明优选将所得合并后样品蒸发浓缩至干,然后使用体积浓度为5%甲醇水溶液溶解,得到分离产物,进行第二次柱层析分离。
[0090]
在本发明中,所述第二次柱层析分离所用填料优选为ods-c18反向柱,玻璃柱规格为5
×
40cm;所述第二次柱层析分离优选包括三个洗脱梯度,洗脱梯度1所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水混合液和甲醇的体积比为95:5,用量优选为250~500ml,更优选为400ml;洗脱梯度2所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水溶液和甲醇的体积比为75:25,用量优选为250~500ml,更优选为400ml;洗脱梯度3所用洗脱剂为体积浓度为0.2%的甲酸水混合液和甲醇,所述甲酸水混合液与甲醇的体积比为50:50,用量优选为250~500ml,更优选为400ml。
[0091]
得到3个洗脱组分后,本发明优选采用高效液相色谱检测法对所述3个洗脱组分进行检测,确定酯型儿茶素-茶氨酸加合物所在组分。在本发明中,所述高效液相色谱检测法的条件优选与上述对萃取相进行高效液相色谱检测确定酯型儿茶素-茶氨酸加合物存在的条件相同,在此不再赘述。
[0092]
本发明优选将酯型儿茶素-茶氨酸加合物所在组分进行高效液相色谱分离,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物。
[0093]
在本发明中,所述高效液相色谱分离所用色谱柱为agilent zorbax sb-aq c18色谱柱,所述色谱柱的尺寸优选为250
×
4.6mm,柱径优选为5μm,流速优选为1ml/min;检测波长为278nm,柱温箱温度为30℃,检测器为dad;流动相包括a相和b相,所述a相为体积浓度为0.2%的甲酸水溶液,b相为甲醇;所述高效液相色谱分离的洗脱条件为:
[0094]
0~4min,5~25%甲酸水溶液;
[0095]
4~7min,25%~38%甲酸水溶液;
[0096]
7~9min,38%~42%甲酸水溶液;
[0097]
9~17min,42%甲酸水溶液;
[0098]
17~19min,42%甲酸水溶液~100%甲醇;
[0099]
19~20min,100%b相~5%甲酸水溶液;
[0100]
20~25min,5%甲酸水溶液。
[0101]
在本发明中,得到具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物(epsfs)后,本发明优选对四种单体化合物进行纯度和质谱鉴定;所述epsfs的检测方法优选为hplc或uplc-ms法,检测流速优选为1ml/min,流动相:a相为体积浓度为0.2%的甲酸水溶液,b相为甲醇,洗脱条件:
[0102]
0~5min,5%甲酸水溶液~20%甲酸水溶液;
[0103]
5~16min,20%甲酸水溶液~25%甲酸水溶液;
[0104]
16~25min,25%甲酸水溶液;
[0105]
25~38min,25%甲酸水溶液~45%甲酸水溶液;
[0106]
38~40min,45%甲酸水溶液~100%甲醇;
[0107]
40~42min,100%甲醇~5%甲酸水溶液;
[0108]
42~55min,5%甲酸水溶液。
[0109]
在本发明中,所述具有式i、式ii、式iii和式iv所示结构的酯型儿茶素-茶氨酸加合物的结构式为
[0110]
[0111][0112]
在本发明中,所述酯型儿茶素-茶氨酸加合物(记为epsfs)中,式i所示结构的化合物中c位置为:
[0113][0114]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0115]
以下实施例中,表没食子儿茶素没食子酸酯和茶氨酸的纯度均为分析纯,纯度分别为95%和99%;
[0116]
以下实施例1~6为不同条件下所得epsfs产率的实施例,将所制备的加合产物直接用甲醇溶解后,采用高效液相色谱检测法进行检测,无需复溶、萃取和柱层析纯化的过程。
[0117]
实施例1
[0118]
不同化学合成反应装置对epsfs含量的影响
[0119]
(1)分别称取50mgegcg(分析级标准品,纯度95%)和50mg茶氨酸(分析级标准品,纯度99%)于反应釜、敞口试管或密闭玻璃瓶中,加入200微升采用浓度为0.01mol/l的氢氧化钠调节ph值为7的水,涡旋3分钟,超声3分钟,然后转移到150℃的烘箱中,加热90分钟,每个样3个重复,得到加合产物;
[0120]
(2)将egcg和茶氨酸的加合产物用纯甲醇溶解,少量多次加入甲醇,直至将产物溶解完全,合并甲醇溶液,定容至5ml;
[0121]
(3)将反应釜、敞口试管和密闭玻璃瓶三种反应装置反应后得到的甲醇溶液分别
进行hplc和uplc-ms检测分析,检测时的流速为1ml/min,流动相:a相为0.2%甲酸水,b相为甲醇,洗脱条件:
[0122]
0~5分钟,5%甲醇水~20%甲醇水;
[0123]
5~16分钟,20%甲醇水-25%甲醇水;
[0124]
16~25分钟,25%甲醇水;
[0125]
25~38分钟,25%甲醇水~45%甲醇水;
[0126]
38~40分钟,45%甲醇水~100%甲醇;
[0127]
40~42分钟,100%甲醇~5%甲醇水;
[0128]
42~55分钟,5%甲醇水;
[0129]
检测分析后的hplc色谱图如图1所示。由图1可知,以反应釜作为反应装置时,得到epsfs的产率最高,密闭玻璃瓶中epsfs的产率次之,敞口试管中epsfs的产率最低。
[0130]
实施例2
[0131]
不同反应温度对epsfs含量的影响
[0132]
(1)分别称取50mgegcg(分析级标准品,纯度95%)和50mg茶氨酸(分析级标准品,纯度99%)于反应釜中,加入200微升采用浓度为0.01mol/l的氢氧化钠调节ph值为7的水,涡旋3分钟,超声3分钟,然后转移到60℃、90℃、120℃、150℃的烘箱中,加热90分钟,每个样3个重复;
[0133]
(2)将egcg和茶氨酸的加合产物用纯甲醇溶液溶解,甲醇溶液溶解时少量多次,将产物溶解完全,合并甲醇溶液,定容至5ml;
[0134]
(3)将不同反应时间的样品通过hplc进行检测分析,检测方法同实施例1。
[0135]
检测分析后的hplc色谱图如图2所示。由图2可知,在不同反应温度下,epsfs含量最高的温度为150℃。
[0136]
实施例3
[0137]
不同反应时间对epsfs含量的影响
[0138]
(1)分别称取50mgegcg(分析级标准品,纯度95%)和50mg茶氨酸(分析级标准品,纯度99%)于反应釜中,加入200微升采用浓度为0.01mol/l的氢氧化钠调节ph值为7的水,涡旋3分钟,超声3分钟,然后转移到150℃的烘箱中,加热30分钟、60分钟、90分钟和180分钟,每个样3个重复;
[0139]
(2)将egcg和茶氨酸的加合产物用纯甲醇溶液溶解,甲醇溶液溶解时少量多次,将产物溶解完全,合并甲醇溶液,定容至5ml;
[0140]
(3)将不同反应时间的样品通过hplc进行检测分析,检测方法同实施例1。
[0141]
检测分析后的hplc色谱图如图3所示。由图3可知,在不同反应温度下,epsfs含量最高的是在150℃下反应90min,随着烘焙时间的延长,epsfs的含量降低。
[0142]
实施例4
[0143]
不同反应液ph值对epsfs含量的影响
[0144]
(1)分别称取50mgegcg(分析级标准品,纯度95%)和50mg茶氨酸(分析级标准品,纯度99%)于反应釜中,加入采用浓度为0.01mol/l的氢氧化钠和盐酸调节ph值后的200微升不同ph值(ph 3、5、7或9)的水,涡旋3分钟,并超声3分钟,然后转移到150℃的烘箱中,加热90分钟,每个样3个重复;
[0145]
(2)将egcg和茶氨酸的加合产物用纯甲醇溶液溶解,甲醇溶液溶解时少量多次,将产物溶解完全,合并甲醇溶液,定容至5ml;
[0146]
(3)将不同反应时间的样品通过hplc进行检测分析,检测方法同实施例1。
[0147]
检测分析后的hplc色谱图如图4所示。由图4可知,在不同ph值下,epsfs含量最高时ph值为9。
[0148]
实施例5
[0149]
不同加水量对epsfs含量的影响
[0150]
(1)分别称取50mgegcg(分析级标准品,纯度95%)和50mg茶氨酸(分析级标准品,纯度99%)于反应釜中,分别加入0、50、200微升采用浓度为0.01mol/l的氢氧化钠调节ph值为9的水,涡旋3分钟,并超声3分钟,然后转移到150℃的烘箱中,加热90分钟,每个样3个重复;
[0151]
(2)将egcg和茶氨酸的加合产物用纯甲醇溶液溶解,甲醇溶液溶解时少量多次,将产物溶解完全,合并甲醇溶液,定容至5ml;
[0152]
(3)将不同加水量反应后样品通过hplc进行检测分析,检测方法同实施例1。
[0153]
检测分析后的hplc色谱图如图5所示。由图5可知,在不同加水量下,epsfs含量最高的是200μl。
[0154]
实施例6
[0155]
不同极性试剂对egcg-茶氨酸加合产物的萃取:
[0156]
将实施例1中采用反应釜制备的egcg-茶氨酸加合产物用纯水(20ml)进行溶解,依次进行涡旋和超声,使加合产物完全溶解混匀,得到待萃取液;
[0157]
分别使用20ml石油醚、二氯甲烷、乙酸乙酯和正丁醇,分别对待萃取液进行萃取,在加入有机试剂进行萃取时,摇匀,静置5min后,用滴管吸取有机层液体,得到有机萃取液,每种萃取剂萃取3次,合并每种萃取剂的3次萃取液,分别得到4个萃取组分样品,再分别将4个萃取组分样品进行旋蒸浓缩至干,用甲醇对4萃取个组分进行溶解,通过hplc进行检测分析,得到不同萃取组分中epsfs含量,具体检测方法同实施例1。
[0158]
采用不同极性试剂对egcg-茶氨酸加合产物的萃取色谱图如图6所示,其中,a为添加萃取剂前的原液的色谱图,b为4个组分的萃取色谱图;由图6可知,只有乙酸乙酯层和正丁醇层中含有epsfs,并且两个萃取层中epsfs含量差异不大,对epsfs萃取效果所用有机试剂为乙酸乙酯或正丁醇。
[0159]
实施例7
[0160]
按照图7所述流程,分别称取4gegcg(分析级标准品,纯度95%)和4g茶氨酸(分析级标准品,纯度99%)于反应釜中,加入4ml调节ph=9的水,500r/min涡旋3分钟,超声3分钟,超声功率为200w,频率为40khz,将装有反应物的反应釜转移到150℃的烘箱中,加热90分钟,进行亲核反应,将所得产物用200ml纯水在40℃加热超声溶解后,转移到500ml分液漏斗中,向所得水溶液中加入200ml乙酸乙酯,摇匀,静置5分钟,进行萃取,有机试剂和水明显出现分层,将分液漏斗下端的开关打开,分别得到水层溶液和乙酸乙酯层溶液;重复以上步骤,重新对水层溶液进行萃取,共萃取3次,合并乙酸乙酯层溶液,旋转蒸发浓缩至干,得到乙酸乙酯层萃取组分;
[0161]
将所得乙酸乙酯层萃取组分用纯甲醇溶液溶解后,将所得甲醇溶液(400mg/ml)与
硅胶混合均匀后于水浴锅中将甲醇挥干,得到待分离样品;将所述待分离样品通过干法上样进行硅胶柱分离;玻璃柱规格为5
×
40cm,洗脱试剂为二氯甲烷、甲醇和甲酸,体积比为30:10:2,共1200ml作为洗脱液进行洗脱,每接100ml流分作为一个组分,将洗脱得到的样品在薄层层析板快速分离,以二氯甲烷/甲醇/甲酸(30:10:2,v/v/v)作为展开剂,将薄层层析板于254nm的紫外灯下观察斑点分布情况,通过硫酸乙醇溶液(质量浓度为5%)进行显色,将薄层层析板上各流分样品显色后的斑点与对照样的斑点进行对比,将具有相同或者相似斑点的流分进行合并,将合并后样品进行蒸发浓缩至干,使用5%甲醇水溶液对样品进行溶解,将所得样品通过ods-c18柱进行分离纯化,c18反向柱分离的洗脱液为甲醇、水和甲酸的混合溶液,洗脱梯度1为0.2%甲酸水:甲醇(v/v,95:5),400ml;洗脱梯度2为0.2%甲酸水:甲醇(v/v,75:25),400ml;洗脱梯度3为0.2%甲酸水:甲醇(v/v,50:50),400ml,在不同洗脱梯度洗脱下,得到洗脱组分1~3;
[0162]
采用高效液相色谱法对3个洗脱组分进行检测,色谱柱为agilent zorbax sb-aq c18色谱柱,流动相包括a相和b相,所述a相为体积浓度0.2%的甲酸水溶液,b相为甲醇;流速为1ml/min,检测波长为278nm,检测器为dad,柱温为30℃,洗脱条件为:
[0163]
0~5min,5%甲酸水溶液~20%甲酸水溶液;
[0164]
5~16min,20%甲酸水溶液~25%甲酸水溶液;
[0165]
16~25min,25%甲酸水溶液;
[0166]
25~38min,25%甲酸水溶液~45%甲酸水溶液;
[0167]
38~40min,45%甲酸水溶液~100%甲醇;
[0168]
40~42min,100%甲醇~5%甲酸水溶液;
[0169]
42~55min,5%甲酸水溶液;
[0170]
根据上述高效液相色谱的检测结果,确定组分1(fr.1)主要成分为没食子酸(ga);组分2(fr.2)主要成分为egcg(表没食子儿茶素没食子酸酯)和gcg(没食子儿茶素没食子酸酯);组分3(fr.3)的主要成分为epsfs,即酯型儿茶素-茶氨酸加合物所在组分。
[0171]
将所述组分3(fr.3)进行高效液相色谱分离,分离色谱柱为agilent zorb ax sb-aq c
18
(250
×
4.6mm,5μm)柱,流速1ml/min,流动相为甲醇(b相)和体积浓度0.2%甲酸水溶液(a相),洗脱条件为:梯度洗脱0~4min,5%甲酸水溶液~25%甲酸水溶液;47min,25%甲酸水溶液~38%甲酸水溶液;7~9min,38%甲酸水溶液-42%甲酸水溶液;9~17min,42%甲酸水溶液;17~19min,42%甲酸水溶液-100%甲醇;19~20min,100%甲醇~5%甲酸水溶液;20~25min,5%甲酸水溶液;检测波长为278nm,柱温箱温度为30℃,检测器为dad,得到四种黄酮生物碱,分别为egcg的c6和c8与茶氨酸strecker降解产物结合形成的8-c-n-ethyl-2-pyrrolidone substituted(-)-epigallocatechin-3-o-gallate(i和ii)和6-c-n-ethyl-2-pyrrolidone substituted(-)-epigallocatech in-3-o-gallate(iii和iv)。
[0172]
将所述组分3进行高效液相色谱分离后epsfs的色谱图如图8所示,图8中的四个epsfs化合物结构(1、2、3、4)分别对应i、ii、iii和iv。
[0173]
epsfs的性状验证:
[0174]
采用hplc或uplc-ms法,对图8得到的四种单体化合物进行纯度和质谱鉴定;检测流速为1ml/min,流动相:a相为体积浓度为0.2%的甲酸水溶液,b相为甲醇,洗脱条件:
[0175]
0~5min,5%甲酸水溶液~20%甲酸水溶液;
[0176]
5~16min,20%甲酸水溶液~25%甲酸水溶液;
[0177]
16~25min,25%甲酸水溶液;
[0178]
25~38min,25%甲酸水溶液~45%甲酸水溶液;
[0179]
38~40min,45%甲酸水溶液~100%甲醇;
[0180]
40~42min,100%甲醇~5%甲酸水溶液;
[0181]
42~55min,5%甲酸水溶液;
[0182]
检测所得式i~iv化合物的特性为:
[0183]
epsfs1(式i)的质谱碎片离子信息见图9,特性如下:
[0184]
1)、白色无定型粉末;
[0185]
2)、uvλ
max
(nm):280;
[0186]
3)、lc-ms(负离子模式):m/z=568.1441([m-h]-,c
28h27
no
12-理论计算值为568.1533)。
[0187]
4)、核磁共振波谱数据见表1。
[0188]
epsfs2(式ii)的质谱碎片离子信息见图10,特性如下:
[0189]
1)、白色无定型粉末;
[0190]
2)、uvλ
max
(nm):280;
[0191]
3)、lc-ms(负离子模式):m/z=568.1449([m-h]-,c
28h27
no
12-理论计算值为568.1533)。
[0192]
4)、核磁共振波谱数据见表1。
[0193]
epsfs3(式iii)的质谱碎片离子信息见图11,特性如下:
[0194]
1)、白色无定型粉末;
[0195]
2)、uvλ
max
(nm):280;
[0196]
3)、lc-ms(负离子模式):m/z=568.1459([m-h]-,c
28h27
no
12-理论计算值为568.1533)。
[0197]
4)、核磁共振波谱数据见表1。
[0198]
epsfs4(iv)的质谱碎片离子信息见图12,特性如下:
[0199]
1)、白色无定型粉末;
[0200]
2)、uvλ
max
(nm):280;
[0201]
3)、lc-ms(负离子模式):m/z=568.1452([m-h]-,c
28h27
no
12-理论计算值为568.1533)。
[0202]
4)、核磁共振波谱数据见表1。
[0203]
表1 egcg-茶氨酸加合物的核磁共振波谱数据
[0204]
[0205][0206]
注:1h nmr和
13
c nmr在600mhz条件下测试,δ单位为ppm,溶剂为氘代甲醇,表格的空白处说明此处无数据。s:单重峰;m:多重峰;d:双重峰;dd:四重峰。
[0207]
由图9~12和表1可知,本发明成功合成并分离纯化得到式i~iv所示结构的酯型儿茶素-茶氨酸加合物epsfs。
[0208]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1