一种基因生物制剂及其制备方法和应用与流程
1.本发明涉及基因编辑、生物细胞技术领域,特别是涉及一种基因生物制剂及其制备方法和应用。
背景技术:
2.近几年来,基因编辑技术发展迅速,尤其是crispr-cas9,crispr(clustered regularly interspaced short palindromicrepeats)规律成簇间隔短回文重复;cas9(crispr associated nuclease)是crispr相关核酸酶,crispr-cas9是最新出现的一种由rna指导的,利用cas9核酸酶对靶向基因进行编辑的技术。2013年2月15日发表在《科学》(science)的两篇文章,证明cas9系统能在293t,k562,ips等多种细胞中,进行有效的靶向酶切,且非同源重组(nhej)及同源重组(hr)各自效率在3-25%之间,重组效率与talen剪切效果相当。文章还证明,多个靶点可以同时进行靶向剪切。这些工作将进一步靶向基因操纵推向高潮,使得多个基因敲除、敲入变得更为简单、高效。
3.经过多年研究,cas9核酸酶对靶向基因进行编辑技术已趋于成熟。可实现对目标基因的定点突变、基因敲除和基因敲入。目前,该技术在动植物育种、干细胞定向分化、遗传疾定点修复等领域得到迅猛发展。
4.一般情形下,crispr-cas9基因敲除技术中,使用一条grna靶向目标序列进行基因剪切,并利用修复时的缺失,进行基因剪切,增加碱基,造成移码突变,导致基因沉默;还有一种情形,使用两条grna靶向目标序列进行基因剪切,造成基因片段缺失,导致基因沉默。但这两种方法均可能出现一个问题:即使造成基因移码或基因片段缺失,但基因仍旧可以正常表达出蛋白的情况,此时基因所表达的蛋白为非原始野生型蛋白,其功能及性状都可能发生改变,这种基因有可能引发其他副作用产生。
技术实现要素:
5.基于此,有必要针对上述问题,提供一种基因敲除插序方法,通过优化方式使目的基因被剪切后产生可翻译蛋白,且敲除基因可以被降解掉并使其无害化。本发明还提供使用基因敲除插序方法制得的基因生物制剂及其应用。
6.本发明提供的技术方案一如下:
7.一种基因敲除插序方法,该方法是在靶细胞的膜表达型蛋白的胞外段或分泌型蛋白中,使用基因编辑技术,敲除表达型蛋白的胞外段或分泌型蛋白的基因序列并在该敲除基因序列位置插入一段in044核酸序列;所述in044核酸序列为:
8.aactgccggaataccggcccctggctgaagaaggtgctgaagtgtaacacacccgaccctagcaagttcttttcccagctg;
9.其中,所述基因编辑技术包括crispr、转座子、talen和zfn技术中的一种或多种。
10.将所述in044核酸序列插入膜表达型蛋白的胞外段基因序列,或将所述in044核酸序列插入分泌型蛋白的任意位置序列,主要是对靶细胞中插序后的基因起到完全沉默及蛋
白完全降解的作用。
11.在其中一个实施例中,基因敲除插序方法中,所述in044核酸序列对应氨基酸序列为:ncrntgpwlkkvlkcntpdpskffsql。
12.上述in044核酸序列是通过合成一段dna序列后对靶细胞进行基因插入的。在其中一个实施例中,所述dna序列是通过直接加入方式插入靶细胞基因序列中的。
13.上述crispr-cas9技术是通过cas9蛋白和grna对靶细胞的基因敲除后插入in044核酸序列的。
14.上述基因编辑技术包括crispr、转座子、talen和zfn技术中的一种或多种,且所述基因编辑技术中的基因或蛋白和grna是通过递送载体转入所述靶细胞中;其中,所述基因编辑技术中的基因或蛋白包括crispr技术中的ca s9蛋白、talen技术中的tale与内切核酸酶foki偶联元件、zfn技术中的锌指蛋白。基因编辑技术中,优选crispr-cas9基因编辑技术。
15.在一实施例中,基因敲除插序方法中,所述cas9蛋白和grna是通过递送载体转入所述靶细胞中;其中,所述递送系统包括慢病毒、逆转录病毒、普通质粒、附加体、纳米递送系统、电转导及转座子中的一种或几种。
16.本发明还提供一种使用上述基因敲除插序方法制备得到的基因生物制剂。
17.本发明还提供一种基因生物制剂在制备、预防以及治疗疾病药物中的应用。
18.本发明提供的基因敲除插序方法及其制得的基因生物制剂,主要有如下优点:
19.1、基因编辑效率高,最高可以达到100%;
20.2、靶细胞的目的基因被编辑后,由于移码突变无法翻译为蛋白及产生蛋白被快速降解,也就杜绝了不明性状的翻译蛋白产生毒性的副作用问题;
21.3、基因编辑作用于膜表达型蛋白及分泌型蛋白,可使得膜表达蛋白的胞外区域被泛素化降解,导致其无法与其他蛋白结合,分泌型蛋白在分泌过程中会被泛素化降解使其无法分泌至细胞外,可保障被编辑蛋白无法产生原有效果;
22.4、本发明提供基因插序方法及基因生物制剂,应用前景广泛,可用于基因编辑、疾病诊断和治疗等各方面。
附图说明
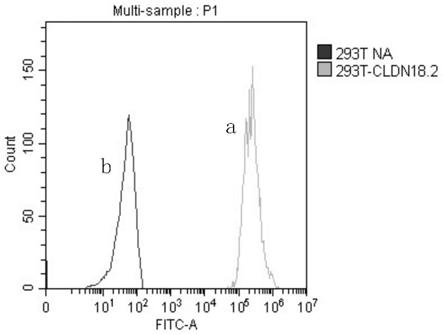
23.图1为实施例1的293t、293t-cldn18.2细胞表达cldn18.2的检测图图2实施例1的ct106、ct090、ct032结构示意图;
24.图3a、3b、3c为实施例2的ct106及ct090的car表达示意图;
25.图4为实施例2的ct106、ct090及对照样nt细胞的扩增曲线;
26.图5为实施例2的ct106和ct090细胞的体外杀伤效果;
27.图6a、6b为实施例2的ct106和ct090细胞的细胞因子释放效果;其中,图6a为il-2细胞因子释放浓度图;图6b为ifn-γ细胞因子释放浓度图;
28.图7a、7b、7c为实施例3的ct106利用grna-pd1敲入in044序列后表面pd-1的表达情况;
29.图8为实施例3的ct106利用grna-pd1敲入in044序列后细胞增殖情况;
30.图9为实施例4的ct032利用grna-antipd1敲入in044序列后,上清中anti-pd1的表
达情况;
31.图10为实施例4的ct032利用grna-antipd1敲入in044序列后细胞增殖情况。
具体实施方式
32.为了便于理解本发明,下面将对本发明进行更全面的描述。
33.本发明提供了一种利基因敲除插序方法,其是在靶细胞的膜表达型蛋白的胞外段或分泌型蛋白中,使用基因编辑技术,敲除细胞膜表达型蛋白的胞外段或分泌型蛋白的基因序列,并在敲除基因序列位置插入一段in044核酸序列。
34.上述in044核酸序列为:aactgccggaataccggcccctggctgaaga aggtgctgaagtgtaacacacccgaccctagcaagttcttttcccagctg。
35.其中,所述基因编辑技术包括crispr、转座子、talen和zfn技术中的一种或多种;基因编辑技术包括crispr、转座子、talen和zfn技术中的一种或多种,且所述基因编辑技术中的基因或蛋白和grna是通过递送载体转入所述靶细胞中;其中,所述基因编辑技术中的基因或蛋白包括crispr技术中的cas9蛋白、talen技术中的tale与内切核酸酶foki偶联元件、zfn技术中的锌指蛋白。
36.本实施例中,基因编辑技术中,优选crispr-cas9基因编辑技术。
37.本发明基因插序法,是将所述in044核酸序列插入膜表达型蛋白的胞外段基因序列中,或将所述in044核酸序列插入分泌型蛋白的任意位置序列中,主要是起到基因完全沉默及蛋白完全降解的功能。
38.上述in044核酸序列对应氨基酸序列为:
39.ncrntgpwlkkvlkcntpdpskffsql。
40.上述in044核酸序列是通过合成一段dna序列后对靶细胞进行基因插入的。在其中一个实施例中,所述dna序列是通过直接加入方式插入靶细胞基因序列中的。
41.上述crispr-cas9技术是通过cas9蛋白和grna对靶细胞的基因敲除后插入in044核酸序列的。
42.在一实施例中,基因敲除插序方法中,所述cas9蛋白和grna是通过递送载体转入所述靶细胞中;其中,基因敲除插序方法中,所述递送系统包括慢病毒、逆转录病毒、普通质粒、附加体、纳米递送系统、电转导及转座子中的一种或几种。
43.本发明还提供一种使用上述基因敲除插序方法制备得到的基因生物制剂。
44.本发明还提供一种基因生物制剂在制备、预防以及治疗疾病药物中的应用。
45.本发明中的基因敲除插序方法,其技术优点如下:
46.1、基因编辑效率高于传统crispr-cas9基因编辑效率。
47.2、目的基因被编辑后,一则因为移码突变无法翻译为蛋白,二则即使产生蛋白也会被快速降解,杜绝了不明性状的翻译蛋白产生的毒性问题,充分保证基因敲除无副产物生产。
48.3、基因编辑作用于膜表达蛋白及分泌型蛋白,可保障被编辑蛋白无法产生作用于细胞外的效果。
49.下面以car-t细胞作为靶细胞为例,对本发明的基因敲除插序方法进行详细说明。
50.本发明中可编辑的靶细胞,即免疫细胞不限于上下文所述的car-t细胞。
cldn18.2、慢病毒包膜质粒pmd2.g(addgene,plasmid#12259)和慢病毒包装质粒pspax2(addgene plasmid#12260)等三个质粒使用lipofectamine3000转入293t中制备慢病毒完整表达载体;分别在48h和72h收集病毒上清,并对收集的病毒上清进行超速离离心浓缩(merck millipore);浓缩后的病毒即可用于感染293t,最终得到过表达cldn18.2的293t细胞系,命名为293t-cldn18.2。
69.如图1中为293t、293t-cldn18.2细胞表达cldn18.2的检测图。图1中,293t-cldn18.2对应右侧曲线(a曲线),表示构建293t-cldn18.2细胞表面的cldn18.2细胞相对于对照样293t对应的左侧曲线(b曲线)为阳性。培养基培养:293t、293t-cldn18.2使用dmem培养基培养。所有培养基均添加10%(v/v)胎牛血清。
70.s300:car结构设计与慢病毒包装
71.cldn18.2-car结构,即靶向cldn18.2的car结构:
72.本发明方法将免疫细胞(car-t)膜上表达第二代car构建在一个表达框上。car的核心结构包括分泌信号肽序列、cd8跨膜区、胞内段刺激信号4-1bb-cd3ζ等,car结构后添加p2a并合成anti-pd1,命名为ct032;car结构后添加p2a并合成绿色荧光蛋白gfp,命名为ct106;在ct106结构基础上,在car结构胞外域插入in044序列,命名为ct090,如图2。
73.如图2所示,cldn18.2 scfv/tm/4-1bb/cd3ζ/p2a/gfp表示免疫细胞的细胞膜上表达的嵌合抗原受体并同时在细胞内合成绿色荧光蛋白gfp,即ct106;而cldn18.2 scfv/in044/tm/4-1bb/cd3ζ/p2a/gfp表示免疫细胞的细胞膜上表达抗原嵌合受体中插入in044序列并同时在细胞内合绿色荧光蛋白gfp,即ct090;cldn18.2 scfv/tm/4-1bb/cd3ζ/p2a/anti-pd1表示免疫细胞的细胞膜上表达的嵌合抗原受体并同时在细胞内合成anti-pd1,即ct032。
74.将表达框克隆至phblv慢病毒载体骨架中,置于ef1α(ef-1α)的启动子下,形成phblv-ef1α-ct032、phblv-ef1α-ct106和phblv-ef1α-ct090,将phblv-ef1α-ct032/ct106/ct090、慢病毒包膜质粒pmd2.g(addgene,plasmid#12259)和慢病毒包装质粒pspax2(addgene plasmid#12260)等三个病毒质粒,使用lipofectamine3000转入293t中制备慢病毒完整表达载体;分别在细胞培养到48h和72h后,转入离心管中离心处理,离心停止后收集病毒上清液,对上清液进行超速离心浓缩(merck millipore);浓缩后的病毒即可用于感染t细胞。
75.s400:car-t细胞制备。
76.4.1慢病毒感染
77.将步骤s100分离纯化的原代t细胞再激活1天后,利用步骤s300包装的3种慢病毒(lv032/lv106/lv107),按moi(1-10)进行慢病毒载体感染,并将感染病毒的t细胞转移至细胞培养瓶,置于37℃,5%co2恒温培养箱中培养。
78.4.2细胞增殖及car阳性率检测
79.t细胞感染后的第6、9、11、13天,每天取样检测细胞数量,第6天分别检测t细胞的car阳性率,每隔1-2天传代补加培养基。
80.利用步骤s300的3种慢病毒载体,成功构建了3种car-t细胞,分别命名为ct032/ct106/ct090,以不感染慢病毒的t细胞为对照(nt)。
81.实施例2
82.本实施例用以说明in044序列插入car结构胞外段对car表达的影响,具体步骤如下:
83.s500:ct106与ct090细胞car阳性率表达情况。
84.图3a、3b、3c分别表nt细胞、ct106及ct090的car阳性率检测图。如图3a、3b、3c所示,以nt细胞作为阴性对照,ct106及ct090细胞绿色荧光蛋白gfp正常表达,说明基因表达正常,gfp表达水平即可认定为基因表达阳性率。但具有in044插入序列的ct090,其car表达完全沉默,证明当在膜表达蛋白胞外区插入in044序列时,该序列所翻译蛋白可介导蛋白降解,实现蛋白表达沉默。
85.s600:ct106与ct090细胞增殖情况。
86.如图4所示,将nt/ct106/ct090细胞增殖情况绘制为生长曲线。以nt细胞和ct106细胞作为对照,ct090细胞的增殖情况与nt、ct106细胞无明显差异,证明in044序列的插入对细胞增殖无显著影响。
87.s700:ct106与ct090细胞体外杀伤情况。
88.5.1、ct106与ct090细胞体外杀伤效率对比
89.将ct106和ct090作为效应细胞,293t和293t-cldn18.2作为靶细胞,按照效靶比1:27、1:9、1:3、1:1比例混合共培养6h后,计算体外杀伤效率,结果如如图5所示,无in044序列插入的ct106细胞发挥正常car-t对293t-cldn18.2阳性靶细胞的杀伤功能,而具有in044序列插入的ct090细胞对293t-cldn18.2阳性靶细胞同nt细胞相同,无特异性杀伤效果。
90.5.2、ct106与ct090细胞体外杀伤上清中细胞因子释放情况检测
91.将ct106和ct090体外杀伤实验上清收集,进行il-2及ifn-γ细胞因子检测,检测结果如图6a、6b所示,无in044序列插入的ct106细胞发挥正常,ct106对293t-cldn18.2阳性靶细胞的具有杀伤功能,并正常进行细胞因子释放,同时ifn-γ细胞因子同阳性靶细胞数量成正比;而具有in044序列插入的ct090细胞对293t-cldn18.2阳性靶细胞同nt细胞相同,无特异性杀伤效果,且基本没有细胞因子释放,证明该in044序列插入后导致car结构被降解基因被敲除。
92.实施例3
93.本实施例用以说明in044序列使用crispr-cas9敲入技术可沉默car-t细胞膜表达pd-1蛋白的表达,具体步骤如下:
94.s800:使用crispr-cas9向ct106细胞中pd1基因敲入in044序列。
95.6.1、序列合成
96.首先合成in044序列,并通过40个pcr循环(聚合酶链式循环反应)和ampure磁珠纯化扩增子。
97.6.2、准备grna-pd1
98.设计并合成靶向人pd-1蛋白胞外区的grna-pd1,将crrna和tracrrna重悬到浓度为200um的idt双工缓冲液中。将crrna和tracrrna解冻溶解,体积1:1混合,98℃孵育5min,在室温中冷却形成100μm grna-pd1溶液。
99.6.3、电转过程
100.第0天:用磁珠分离t细胞,其中,t细胞:磁珠=1:3;并添加il-2细胞因子,加入量为100iu/ml;
101.第2天:脱珠,将细胞放到锥形管中放回37℃培养箱;另外,预热一些opt5+il2培养基,以供后续使用;
102.准备rnp+cas9
103.将rnp与cas9蛋白(5ug/ul)混合,最终体积比grna:pga:cas9,共1:0.8:1.6。室温孵育15min,孵育期间,细胞进行室温离心,即90g,10min。用p3溶液重悬细胞(p3:supplement=9:2),每20ul溶液1.5*10^6~2*10^6个/mlrnp与hdr孵育1min,在每个rnp+hdr管中加入p3重悬细胞(16孔,20ul),混合均匀。将rnp混合物转移到试皿中,使用eh-115程序进行电穿孔处理。电穿孔结束后,立即加入80ul预热过的pbs,放入37℃中,再放15min。用200ul枪将处理过的细胞转移到适当的板中,并用预热的旧培养基进行补液(敲入时,细胞密度不应小于1*106个/ml)。放入37℃培养箱进行培养。
104.第5天:检测外源性基因的表达
105.6.4检测ct106细胞pd1基因敲除前后表达情况
106.如图7a、7b、7c所示,ct106在使用crispr-cas9技术插入in044序列至pd-1基因胞外段后,ct106-in044细胞表面pd1表达完全沉默,可以证明该敲除方法可有效沉默细胞膜表达蛋白。如图8所示,ct106细胞与ct106-in044细胞在增殖能力上无显著差异。
107.实施例4
108.本实施例用以说明in044序列使用crispr-cas9敲入技术可沉默car-t细胞分泌anti-pd-1蛋白的表达,具体步骤如下:
109.s900:使用crispr-cas9技术敲入in044序列至ct032细胞的anti-pd1基因中。
110.合成grna-antipd1,利用s800中的crispr-cas9技术步骤将in044序列敲入ct032细胞的anti-pd1基因中。结果图9所示,ct032细胞中antipd1重组抗体蛋白正常分泌,而接受过crispr-cas9敲入in044的ct032-in044细胞上清中已检测不到anti-pd1重组抗体蛋白表达。如图10所示,ct032细胞与ct032-in044细胞在增殖能力上无显著差异。证明本发明中的技术可有效沉默分泌型蛋白基因表达,并对被编辑细胞无显著影响。
111.本发明提供的基因编辑方法,可有效沉默被编辑基因,使得膜表达蛋白和分泌型蛋白基因完全沉默。
112.以上所述实施例仅表达了本发明的一种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1