一种苉二酰亚胺衍生物及其制备方法和应用
1.本发明涉及有机光电器件材料技术领域,尤其是涉及一种苉二酰亚胺衍生物及其制备方法和应用。
背景技术:
2.基于有机半导体材料的有机场效应晶体管器件具有价廉、质轻、可溶液法加工等特点,是近年来兴起的一种新型电子器件。得益于有机材料本身良好的机械柔性,有机场效应晶体管可广泛应用于柔性可穿戴电子器件上,这是传统的硅基场效应晶体管无法比拟的。目前有机场效应晶体管的迁移率已超过无机非晶硅的水平,且基于有机场效应晶体管的无线射频标签,柔性气体传感器、柔性压力传感器件也相继被报道并受到人们的广泛关注。
3.根据载流子性质的不同,可将有机半导体材料分为p型和n型两种,p型有机半导体制备的场效应晶体管器件中,主要载流子为空穴。目前,p型有机半导体材料发展较为迅速,基于并五苯、红荧烯等分子的器件已经展现出超10cm2v-1
s-1
的迁移率。然而n型有机半导体的研究进展缓慢,其器件迁移率远小于p型材料,主要原因是n型有机半导体的能级调控较为困难,影响了材料和电极之间的载流子传输。同时较高的lumo能级使得n型有机场效应晶体管的性能容易受到大气气氛的损害。
4.因此,开发新型n型有机半导体材料是推动n型有机半导体器件向前发展的重要途径。
技术实现要素:
5.本发明的目的在于克服上述技术不足,提出一种苉二酰亚胺衍生物及其制备方法和应用,解决现有技术中n型有机半导体器件迁移率低的技术问题。
6.为达到上述技术目的,本发明的第一方面提供一种苉二酰亚胺衍生物的结构式如下:
[0007][0008]
通式a中,r1为碳数1~30的烷基,r2为氢、卤素或氰基。
[0009]
本发明的第二方面提供一种苉二酰亚胺衍生物的制备方法,包括如下步骤:
[0010]
将原料a和反式-1,2双(三丁基锡)-乙烯在pd(pph3)4的作用下,在甲苯中进行第一回流反应,经分离纯化,得到中间体b;
[0011]
将中间体b在碘单质的催化作用下,在甲苯中进行光环化反应,经分离纯化,得到
苉二酰亚胺衍生物,合成工艺如下:
[0012][0013]
其中,r1为碳数1~30的烷基。
[0014]
本发明的第三方面提供一种苉二酰亚胺衍生物的应用,本发明第一方面提供的苉二酰亚胺衍生物作为n型半导体应用于有机光电器件。
[0015]
本发明的第四方面提供一种有机场效应晶体管器件,该有机场效应晶体管器件包括本发明第一方面提供的苉二酰亚胺衍生物。
[0016]
与现有技术相比,本发明的有益效果包括:
[0017]
本发明通过向苉基础单元中引入酰亚胺结构,降低材料lumo能级,增加其在空气中的稳定性,开发一种新型构筑模块——苉二酰亚胺,并以此为构筑单元,通过引入吸电子基团(卤素、氰基等),设计并合成一批新型n-型半导体材料,该新型n-型半导体材料能够作为n型半导体应用于有机光电器件。
附图说明
[0018]
图1是本发明实施例1制得的化合物1的氢谱图;
[0019]
图2是本发明实施例2制得的化合物2的氢谱图;
[0020]
图3是本发明实施例3制得的化合物6的氢谱图;
[0021]
图4是本发明实施例4制得的化合物10的氢谱图;
[0022]
图5是本发明实施例6制得的化合物12的氢谱图;
[0023]
图6是本发明实施例7测得的紫外可见(uv-vis)吸收光谱图;
[0024]
图7是本发明实施例7测得的循环伏安曲线;
[0025]
图8是本发明实施例8制得的ofet器件转移曲线图。
具体实施方式
[0026]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0027]
本发明的第一方面提供一种苉二酰亚胺衍生物,该苉二酰亚胺衍生物的结构式如下:
[0028][0029]
通式a中,r1为碳数1~30的烷基,进一步为碳数6~15的烷基,更进一步为正辛基或7-十三烷基,r2为氢、卤素或氰基,进一步为氢、溴或氰基。在本发明的一些优选实施方式中,r2不全为氢。
[0030]
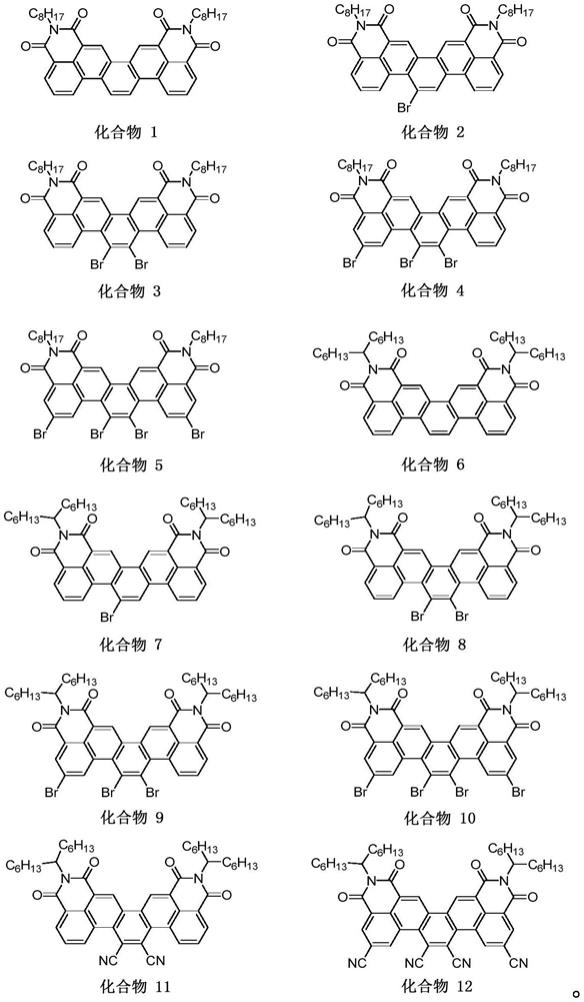
以下表示通式ⅰ所示的化合物的具体实例,但本发明的化合物并不限定于这些化合物。
[0031]
[0032]
本发明的第二方面提供一种苉二酰亚胺衍生物的制备方法,包括如下步骤:
[0033]
s1、将原料a和反式-1,2双(三丁基锡)-乙烯在pd(pph3)4的作用下,在甲苯中进行第一回流反应,经分离纯化,得到中间体b;其中,原料a、反式-1,2双(三丁基锡)-乙烯、pd(pph3)4的摩尔比为1:(0.4~0.6):(0.01~0.05);原料a与甲苯的用量比为1g:(10~30)ml;第一回流反应在氮气保护下进行,第一回流反应的温度为100~130℃,进一步为110℃,时间为10~14h,进一步为12h。
[0034]
s2、将中间体b在碘单质的催化作用下,在甲苯中进行光环化反应,经分离纯化,得到苉二酰亚胺衍生物。其中,中间体b与碘单质的摩尔比为1:(0.3~0.6);中间体b与甲苯的用量比为1g:(200~300)ml;光环化反应在流动光反应器中进行,光源为可见光,光源功率为300~2000w,反应时间为4~8h。
[0035]
合成工艺如下:
[0036][0037]
其中,r1为碳数1~30的烷基,进一步为碳数6~15的烷基,更进一步为正辛基或7-十三烷基。
[0038]
在本发明的一些具体实施方式中,步骤s1和s2具体为:
[0039]
将原料a和pd(pph3)4加入到250ml两口瓶中,氮气保护下用注射器加入100ml无水无氧的甲苯和反式-1,2双(三丁基锡)-乙烯,110℃回流12h;溶液冷却,用真空旋蒸蒸发仪除去甲苯,粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到黄色固体中间体b;
[0040]
将中间体b溶于800ml甲苯中,加入500mg i2于1l圆底瓶中;反应混合溶液被吸入流动光反应器中(配备4个450w汞灯);将反应混合物反复泵入流动光反应器,保持时间为6h;用真空旋转蒸发仪除去溶剂,粗产物用甲醇洗涤以除去过量的i2,以石油醚/二氯甲烷为洗脱剂,用硅胶柱纯化粗土黄固体,用二氯甲烷/甲醇重结晶,最终得到苉二酰亚胺衍生物。
[0041]
进一步地,上述苉二酰亚胺衍生物的制备方法,还包括如下步骤:
[0042]
s3、将化合物ⅰ和n-溴代丁二酰亚胺在浓硫酸的催化作用下进行溴代反应,经分离纯化,得到苉二酰亚胺衍生物。其中,化合物ⅰ、n-溴代丁二酰亚胺、浓硫酸的摩尔比为1:(1.8~2.2):(100~200);溴代反应在室温条件下进行,反应时间为2~4h。该过程中,需要说明的是,通过分离纯化过程可以得到多种不同的溴代产物。
[0043]
合成工艺如下:
[0044][0045]
其中,r1为碳数1~30的烷基,进一步为碳数6~15的烷基,更进一步为正辛基或7-十三烷基;r
21
为氢或溴,且r
21
不全为氢。
[0046]
在本发明的一些具体实施方式中,步骤s3具体为:将化合物ⅰ和n-溴代丁二酰亚胺置于100ml圆底烧瓶中,加入5ml浓硫酸,室温搅拌3h;将反应混合物倒入冰水浴中,用dcm将有机物萃取出来,有机层用无水na2so4干燥,用真空旋转蒸发仪除去溶剂;粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂;用二氯甲烷/甲醇重结晶,最终得到苉二酰亚胺衍生物。
[0047]
进一步地,上述苉二酰亚胺衍生物的制备方法,还包括如下步骤:
[0048]
s4、将化合物ⅱ和氰化亚铜在有机溶剂中进行第二回流反应,经分离纯化,得到苉二酰亚胺衍生物。其中,化合物ⅱ与氰化亚铜的摩尔比为1:(20~40);有机溶剂为n,n-二甲基甲酰胺(dmf)或二甲苯,化合物ⅱ与有机溶剂的用量比为1g:(80~120)ml;第二回流反应在氮气的条件下进行,第二回流反应的温度为140~160℃,时间为12~48h。该过程中,还可根据需要加入钯催化剂和有机膦配体,进一步地,钯催化剂为pd2(dba)3,有机膦配体为1,1'-双(二苯基膦)二茂铁(dppf);化合物ⅱ与钯催化剂、有机膦配体的摩尔比为1:(0.1~0.5):(0.1~0.5)。
[0049]
合成工艺如下:
[0050][0051]
其中,r1为碳数1~30的烷基,进一步为碳数6~15的烷基,更进一步为正辛基或7-十三烷基;r
21
为氢或溴,且r
21
不全为氢;r
22
为氢或氰基,且r
22
不全为氢。
[0052]
在本发明的一些具体实施方式中,步骤s4具体为:将化合物ⅱ和氰化亚铜加入到100ml两口瓶中,氮气保护下用注射器加入20ml无氧的有机溶剂,140~160℃回流12~48h;溶液冷却,除去有机溶剂,粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到苉二酰亚胺衍生物。该过程中,还可根据需要加入钯催化剂和有机膦配体。
[0053]
本发明的第三方面提供一种苉二酰亚胺衍生物的应用,本发明第一方面提供的苉二酰亚胺衍生物作为n型半导体应用于有机光电器件,特别是有机场效应晶体管(ofets)。
[0054]
本发明的第四方面提供一种有机场效应晶体管器件,该有机场效应晶体管器件包括本发明第一方面提供的苉二酰亚胺衍生物。
[0055]
实施例1:化合物1的合成方法
[0056]
化合物1的结构如下:
[0057][0058]
上述化合物1的合成方法,按照如下合成方式:
[0059]
(1)将原料1a(5g,0.0129mol)和pd(pph3)4(250mg)加入到250ml两口瓶中,氮气保护下用注射器加入100ml无水无氧的甲苯和反式-1,2双(三丁基锡)-乙烯(3.90g,0.0064mol),110℃回流12h。溶液冷却,用真空旋蒸蒸发仪除去甲苯,粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到黄色固体中间体1b(3.44g,83%)。
[0060]
(2)将中间体1b(3g,0.0047mol)溶于800ml甲苯中,加入500mg i2于1l圆底瓶中。反应混合溶液被吸入流动光反应器中(配备4个450w汞灯)。将反应混合物反复泵入流动光反应器,保持时间为6h。用真空旋转蒸发仪除去溶剂,粗产物用甲醇洗涤以除去过量的i2。以石油醚/二氯甲烷为洗脱剂,用硅胶柱纯化粗土黄固体。用二氯甲烷/甲醇重结晶,最终得到黄色固体化合物1(2.72g,91%)。化合物1具体氢谱图见图1。
[0061]
具体反应方程式如下:
[0062][0063]
实施例2:化合物2、3、4、5的合成方法
[0064]
化合物2、3、4、5的结构如下:
[0065][0066]
上述化合物2、3、4、5的合成方法,按照如下合成方式:
[0067]
(3)将化合物1(400mg,0.6242mmol)和n-溴代丁二酰亚胺(222.19mg,1.2484mmol)置于100ml圆底烧瓶中,加入5ml浓硫酸,室温搅拌3h。将反应混合物倒入冰水浴中,用dcm将有机物萃取出来,有机层用无水na2so4干燥,用真空旋转蒸发仪除去溶剂;粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂;用二氯甲烷/甲醇重结晶,最终得到黄色固体化合物
2、3、4、5(2:35mg、3:30mg、4:37mg、5:38mg)。其中,化合物2具体氢谱图见图2,化合物2、3、4、5的反应方程式如下:
[0068][0069]
实施例3:化合物6的合成方法
[0070]
上述化合物6的结构如下:
[0071][0072]
上述化合物6的合成方法,按照如下合成方式:
[0073]
(1)将原料6a(5g,0.0109mol)和pd(pph3)4(250mg)加入到250ml两口瓶中,氮气保护下用注射器加入100ml无水无氧的甲苯和反式-1,2双(三丁基锡)-乙烯(3.31g,0.0055mol),110℃回流12h。溶液冷却,用真空旋蒸蒸发仪除去甲苯,粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到黄色固体中间体6b(3.64g,85%)。
[0074]
(2)将中间体6b(3g,0.0038mol)溶于800ml甲苯中,加入500mg i2于1l圆底瓶中。反应混合溶液被吸入流动光反应器中(配备4个450w汞灯)。将反应混合物反复泵入流动光反应器,保持时间为6h。用真空旋转蒸发仪除去溶剂,粗产物用甲醇洗涤以除去过量的i2。以石油醚/二氯甲烷为洗脱剂,用硅胶柱纯化粗土黄固体。用二氯甲烷/甲醇重结晶,最终得到黄色固体化合物6(2.81g,94%)。化合物6具体氢谱图见图3。
[0075]
具体反应方程式如下:
[0076][0077]
实施例4:化合物7、8、9、10的合成方法
[0078]
化合物7、8、9、10的结构如下:
[0079][0080]
上述化合物7、8、9、10的合成方法,按照如下合成方式:
[0081]
(3)将化合物6(400mg,0.5122mmol)和n-溴代丁二酰亚胺(182.28mg,1.0244mmol)置于100ml圆底烧瓶中,加入5ml浓硫酸,室温搅拌3h。将反应混合物倒入冰水浴中,用dcm将有机物萃取出来,有机层用无水na2so4干燥,用真空旋转蒸发仪除去溶剂;粗产物经硅胶层析纯化,以石油醚/二氯甲烷为洗脱剂;用二氯甲烷/甲醇重结晶,最终得到黄色固体化合物7、8、9、10(7:37mg;8:32mg;9:35mg;10:38mg)。化合物10具体氢谱图见图4。
[0082]
反应方程式如下:
[0083][0084]
实施例5:化合物11的合成方法
[0085]
化合物11的结构如下:
[0086][0087]
上述化合物11的合成方法,按照如下合成方式:
[0088]
(4)将化合物8(200mg,0.2130mmol)和氰化亚铜(381.56mg,4.260mmol)加入到100ml两口瓶中,氮气保护下用注射器加入20ml无氧的n,n-二甲基甲酰胺(dmf),150℃回流12h。溶液冷却,用真空旋蒸蒸发仪除去dmf,粗产物经硅胶层析纯化,以石油醚/二氯甲烷为
洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到黄色固体化合物11(74mg)。反应方程式如下:
[0089][0090]
实施例6:化合物12的合成方法
[0091]
化合物12的结构如下:
[0092][0093]
(4)将化合物10(200mg,0.1823mmol)、氰化亚铜(653.32mg,7.295mmol)、pd2(dba)3(33.40mg,0.0365mmol)和dppf(20.22mg,0.0365mmol)加入到100ml两口瓶中,氮气保护下用注射器加入20ml无氧的二甲苯,140℃回流48h。溶液冷却,粗产品经硅胶层析,以石油醚为洗脱剂除去二甲苯,以石油醚/二氯甲烷为洗脱剂洗脱;再用二氯甲烷/甲醇重结晶,得到黄色固体化合物12(15mg)。化合物12具体氢谱图见图5。反应方程式如下:
[0094][0095]
实施例7:光电性能测试
[0096]
将化合物12溶于氯仿中,用uv-1800型紫外-可见分光光度计测试其紫外可见吸收光谱图,见图6。根据起始吸收波长λ
onset
和能隙公式:e
gopt
=1240/λ
onset
计算得到e
gopt
=2.88ev。
[0097]
将化合物12溶于二氯甲烷中,以四丁基六氟磷酸铵为电解质;采用三电极体系,以ag/ag
+
(1mol/l)电极为参比电极,以铂丝电极为对电极,以铂碳电极为工作电极;用chi660e型电化学工作站测试其循环伏安曲线,扫速50mv/s,测试区间-2v—2v,循环3次,灵敏度10-5
a/v,结果见图7。根据公式:e
lumo
=-(e
red-e
(fcfc+)
+4.8)(e
red
为循环伏安曲线测得的化合物12还原电势;e
(fc/fc+)
为二茂铁的氧化还原电势)计算出lumo能级e
lumo
=-4.00ev;根据e
gopt
和e
lumo
可计算出相应的homo能级e
homo
=-6.88ev。
[0098]
实施例8:制备器件ofet
[0099]
本实施例提供了一种制备ofet的方法,包括如下步骤:
[0100]
(1)将含有300nm氧化层的重掺杂硅片(掺杂磷的硅片,面积为1.3平方厘米)置于含清洁剂的水溶液中超声清洗15分钟,随后在去离子水中清洗10分钟,最后分别用丙酮和
异丙醇超声清洗15分钟,将干净的硅片用氮气吹干备用;
[0101]
(2)用浓硫酸和双氧水(7:3,v/v)配制食人鱼溶液,将干净的硅片置于食人鱼溶液中浸泡30分钟,随后用去离子水超声清洗若干次,并用氮气枪吹干备用;
[0102]
(3)将处理后的硅片置于培养皿中,滴入一滴十八烷基三氯硅烷(ots),随后用120℃真空干燥处理2小时;将ots修饰后的硅片先后用甲苯和异丙醇超声清洗15分钟,并吹干备用;
[0103]
(4)将化合物12溶于氯仿中配置成浓度为5mg/ml的溶液,随后使用旋涂法在ots修饰的硅片上制备化合物12的薄膜,旋涂速度3000rpm,时间为60s;将涂有化合物12薄膜的硅片置于加热板上加热10分钟,温度为120℃;
[0104]
(5)将含有电极图案的掩模版覆盖在上述涂有化合物12薄膜的硅片上,并放于真空腔内抽真空至5
×
10-4
pa,采用0.01nm/s的速率在化合物12薄膜上蒸镀30nm厚的金电极,得到ofet器件。
[0105]
器件测试:实施例8中的ofet器件测试使用的仪器为吉时利4200scs半导体分析仪。测试时在ofet漏极施加70v的偏置电压,在栅极施加0到70v的扫描电压,此时在ofet沟道中诱导出的载流子种类为电子,该种条件下测试得到的器件迁移率为电子的迁移率,也称为n型迁移率,为0.018cm2v-1
s-1
。
[0106]
评价结果:ofet的开关比105,电子迁移率为0.018cm2v-1
s-1
,转移曲线见图8。
[0107]
本发明所设计并合成的新型有机光电材料苉二酰亚胺衍生物合成路线简单、产率高、可大量合成;相对于苉,引入两个酰亚胺基团和溴来调节分子的lumo能级能够使其更好地应用于有机光电器件中,特别是有机场效应晶体管;该新型有机光电材料苉二酰亚胺衍生物具有化学稳定性高、溶解性好的优点;应用化合物12所制备的ofet器件显示出较高的电子迁移率,呈现出优异的性能;并且,酰亚胺基团中的烷基链增强了分子的溶解度。
[0108]
以上所述本发明的具体实施方式,并不构成对本发明保护范围的限定。任何根据本发明的技术构思所做出的各种其他相应的改变与变形,均应包含在本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1