一种新的光标记试剂及其在细胞表面蛋白质组和N-糖基化富集分析中的应用
一种新的光标记试剂及其在细胞表面蛋白质组和n-糖基化富集分析中的应用
技术领域
1.本发明属于分析化学领域,具体地,本发明涉及一种新的光标记试剂及其在细胞表面蛋白质组和n-糖基化富集分析中的应用,更具体地,所述新的光标记试剂为c16-mbp。
背景技术:
2.细胞表面蛋白质组是由生物体中25%的蛋白质编码基因编码的一类蛋白质,在调节细胞与周围环境的通信中起着关键作用。许多膜上蛋白质又发生糖基化,它们在许多细胞功能和活动中发挥着关键作用,如细胞间相互作用、病原体识别、离子转运和信号转导。这些蛋白包括许多细胞表面受体、离子通道和转运蛋白,它们约占fda批准的所有药物靶点的70%,因此可以反映细胞表面蛋白的药理相关性。此外,细胞表面蛋白的糖基化对细胞粘附、迁移和免疫反应调节至关重要。异常蛋白糖基化被认为是癌症的标志和免疫治疗的重要靶点。因此,对细胞表面蛋白及其糖基化的全面分析可能有助于更好地理解它们在各种细胞活动和疾病发展中的功能,从而帮助发现新的生物标志物和药物靶点。然而,与细胞内蛋白相比,大多数膜蛋白的自然表达水平很低。其高特异性纯化的困难进一步阻碍了对其结构和功能的了解。
3.为了有效地分离细胞表面蛋白,近年来广泛报道的方法大致可分为两大类,一类依赖于细胞表面蛋白的理化特性,另一类依赖于细胞表面蛋白的标记。与依赖理化性质分离的高速离心法和基于辅助溶解的去垢剂相比,采用酰肼化学、“点击化学”和细胞表面多糖的酶标记方法虽然成功鉴定了数百种细胞表面蛋白,但是由于现有方法的细胞表面靶向能力有限,其蛋白质组覆盖率和选择性仍不理想。此外,化学/酶标记可能会改变聚糖的组成和结构,从而导致质谱分析(ms分析)难以完整地阐明聚糖/糖肽。虽然上述限制可以通过细胞表面蛋白质及其完整糖肽的串联富集得到部分解决,但是多重富集过程中样品的损失可能会严重降低鉴定规模。
技术实现要素:
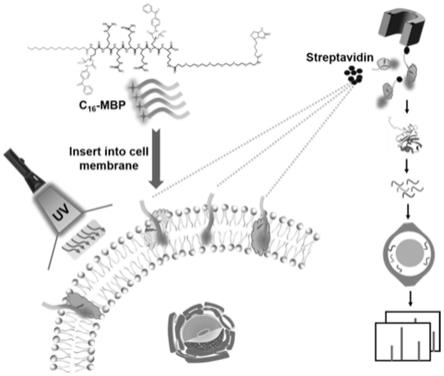
4.鉴于此,为了克服目前本领域存在的上述技术问题,本发明的目的在于提供一种新的光标记试剂,以实现高效标记和方便富集细胞表面蛋白及其糖基化的目的。通过棕榈酸的膜靶向功能以及4-(n-马来酰亚胺)二苯甲酮(mbp)基团与细胞膜上探针临近蛋白的共价结合,实现c16-mbp试剂对紫外交联的细胞膜表面蛋白及其糖基化的大规模富集,所述c16-mbp试剂主要包括四个部分,分别是膜靶向基团、七肽肽链基团、生物素基团和光交联基团。
5.本发明的上述目的通过以下技术方案得以实现:
6.本发明的第一方面提供了一种新型的光标记试剂c16-mbp。
7.进一步,所述光标记试剂c16-mbp的结构式如式i所示:
[0008][0009]
进一步,所述新型的光标记试剂c16-mbp一部分为棕榈酸,具有细胞膜靶向的功能,一部分包含生物素基团,可以特异性地与商业化的链霉亲和素修饰磁珠结合,所述新型的光标记试剂c16-mbp还包括二苯甲酮基团,使其在365nm波长紫外光照下与临近的膜蛋白发生共价反应,从而实现细胞膜表面蛋白的特异性富集。
[0010]
在本发明中,所述“c16-mbp”,同“新型的光标记试剂”、“新型的光标记试剂c16-mbp”、“c16-mbp试剂”、“光标记试剂c16-mbp”、“光标记试剂”、“细胞膜定位探针”、“细胞膜定位探针c16-mbp”、“化合物1”,是指本发明实施例中合成的一种新型的光标记试剂,在该试剂中含有光交联基团、生物素基团和棕榈酸膜靶向基团,可应用于细胞表面蛋白质组和n-糖基化富集鉴定中。
[0011]
本发明的第二方面提供了一种本发明第一方面所述的新型的光标记试剂c16-mbp的制备方法。
[0012]
进一步,所述方法包括如下步骤:
[0013]
(1)将c16-crrrrck-peg6-biotin和4-(n-马来酰亚胺基)二苯甲酮分别溶解于pbs溶液中并混合得到混合溶液;
[0014]
(2)在冰水浴中对步骤(1)中所述的混合溶液进行超声处理,得到粗产物;
[0015]
(3)将步骤(2)中所述的粗产物用脱盐柱进行纯化,收集纯化后的馏分经干燥得到本发明第一方面所述的新型的光标记试剂c16-mbp;
[0016]
优选地,所述c16-crrrrck-peg6-biotin的结构式如式ⅱ所示:
[0017][0018]
在本发明中,所述化合物2为c16-crrrrck-peg6-biotin,简称c16-pep-biotin,由本发明自行设计合成得到;所述化合物3为4-(n-马来酰亚胺基)二苯甲酮,来源于sigma-aldrich,cas#:92944-71-3。
[0019]
进一步,步骤(1)中所述c16-crrrrck-peg6-biotin的用量为20mg、11.25mm;
[0020]
优选地,步骤(1)中所述4-(n-马来酰亚胺基)二苯甲酮的用量为6.24mg、22.50mm;
[0021]
优选地,步骤(1)中所述pbs溶液的用量为1ml;
[0022]
优选地,步骤(2)中所述超声处理的方式为脉冲超声,1s开,1s关;
[0023]
优选地,步骤(2)中所述超声处理的时间为30min;
[0024]
优选地,步骤(3)中所述干燥的方式为冷冻干燥。
[0025]
本发明的第三方面提供了本发明的第一方面所述的新型的光标记试剂c16-mbp在细胞膜表面蛋白及其糖基化富集分析中的应用。
[0026]
进一步,在使用365nm波长紫外光照下新型的光标记试剂c16-mbp与细胞膜蛋白之间发生共价偶联,进而通过链霉亲和素修饰磁珠与c16-mbp中的生物素基团的亲和作用实现各种细胞膜表面蛋白的选择性和广谱性富集。
[0027]
本发明的第四方面提供了本发明第一方面所述的新型的光标记试剂c16-mbp在细胞膜表面蛋白及其糖基化富集的质谱鉴定中的应用。
[0028]
本发明的第五方面提供了一种基于本发明第一方面所述的新型的光标记试剂c16-mbp的细胞膜表面蛋白及其糖基化富集分析方法。
[0029]
进一步,所述方法包括如下步骤:
[0030]
1)将待测细胞样品用pbs溶液清洗后,加入本发明第一方面所述的新型的光标记试剂c16-mbp进行孵育;
[0031]
2)孵育结束后弃去溶液,pbs溶液清洗后,进行紫外光交联;
[0032]
3)紫外光交联后,收集细胞,离心去除上清,加入ripa裂解液裂解细胞,超声处理后,离心取上清;
[0033]
4)在步骤3)所述的上清中加入链霉亲和素修饰磁珠进行孵育,孵育后,磁性分离弃去上清,ripa裂解液、pbs溶液分别清洗,磁性分离弃去上清得到捕获纯化的细胞膜表面蛋白的磁珠;
[0034]
5)在步骤4)得到的磁珠中加入β-巯基乙醇和上样缓冲液,变性后离心取上清进行sds-聚丙烯酰胺凝胶电泳。
[0035]
进一步,步骤1)中所述新型的光标记试剂c16-mbp的终浓度为10-50μm;
[0036]
优选地,步骤1)中所述新型的光标记试剂c16-mbp的终浓度为20μm;
[0037]
优选地,步骤1)中所述孵育的条件为4℃、10-30min;
[0038]
更优选地,步骤1)中所述孵育的条件为4℃、20min;
[0039]
优选地,步骤2)中所述紫外光交联的条件为紫外光波长365nm、功率150w、照射时间1-5min;
[0040]
更优选地,步骤2)中所述紫外光交联的条件为紫外光波长365nm、功率150w、照射时间1min;
[0041]
优选地,步骤3)中所述离心去除上清的离心条件为1000g、3min;
[0042]
优选地,步骤3)中所述ripa裂解液的用量为350-450μl;
[0043]
更优选地,步骤3)中所述ripa裂解液的用量为400μl;
[0044]
优选地,步骤3)中所述超声处理的方式为脉冲超声,2s开,2s关;
[0045]
优选地,步骤3)中所述超声处理的时间为5min;
[0046]
优选地,步骤3)中所述离心取上清的离心条件为16000g、10min;
[0047]
优选地,步骤4)中所述链霉亲和素固载磁珠的用量为50μl;
[0048]
优选地,步骤4)中所述孵育的条件为4℃、1h;
[0049]
优选地,步骤5)中所述β-巯基乙醇的用量为2μl;
[0050]
优选地,步骤5)中所述上样缓冲液的用量为10μl;
[0051]
优选地,步骤5)中所述变性的条件为95℃、10min。
[0052]
本发明的第六方面提供了一种基于本发明第一方面所述的新型的光标记试剂c16-mbp的细胞膜表面蛋白及其糖基化富集的质谱鉴定方法。
[0053]
进一步,所述方法包括如下步骤:
[0054]
①
实验组为采用本发明第五方面所述方法的步骤4)中得到的磁珠中依次加入ripa裂解液、pbs溶液、nh4hco3溶液清洗,通过清洗步骤去除磁珠上的非特异性吸附,对照组为加入不含c16的探针;
[0055]
②
清洗后磁性分离弃去上清,进行胰蛋白酶酶解,磁性分离后弃去磁珠获取上清液及其中的酶解产物,酶解产物经过脱盐小柱脱盐得到洗脱液;
[0056]
③
对步骤
②
得到的洗脱液进行质谱定量分析,比较实验组和对照组所鉴定蛋白的定量差异,卡值扣除非特异性吸附的非膜蛋白后,即可得到高置信度的细胞膜表面相关蛋白;采用高场非对称波形离子迁移谱技术在不富集n-糖肽的胰蛋白酶消化肽中直接鉴定n-糖基化蛋白。
[0057]
进一步,步骤
①
中所述ripa裂解液、pbs溶液、nh4hco3溶液的用量均为200μl;
[0058]
优选地,步骤
②
中所述胰蛋白酶酶解包括如下步骤:加入二硫苏糖醇水浴中还原,还原后加入碘乙酰胺静置进行烷基化处理得到变性后的蛋白,在变性后的蛋白中加入胰蛋白酶水浴中进行孵育;
[0059]
更优选地,所述二硫苏糖醇的终浓度为10mm;
[0060]
更优选地,所述加入二硫苏糖醇水浴的条件为56℃、1h;
[0061]
更优选地,所述碘乙酰胺的终浓度为50mm;
[0062]
更优选地,所述静置的条件为暗处、30min;
[0063]
更优选地,所述胰蛋白酶的用量为1μg;
[0064]
更优选地,所述加入胰蛋白酶水浴的条件为37℃、12h;
[0065]
优选地,所述脱盐小柱为c18 zip-tips脱盐小柱。
[0066]
本发明提供的方法流程和原理如下:培养皿中的待测细胞加入c16-mbp试剂孵育后,使用365nm波长紫外照射,促使c16-mbp试剂中的二苯甲酮基团与临近蛋白形成共价键,从而实现细胞表面的生物素化。然后收集细胞将细胞超声裂解所得样品与链霉亲和素修饰磁珠孵育,清洗除去非特异性吸附的蛋白与其他分子,即可实现细胞表面膜蛋白的特异性富集。后续结合高场非对称波形离子迁移谱(faims)技术,在不进行串联n-糖肽富集的情况下,直接从富集的膜蛋白中分析相应的糖基化蛋白。
[0067]
进一步,为了获得更为准确可靠的细胞表面膜蛋白鉴定结果,本发明采用无c16即无膜靶向功能的探针与细胞孵育,之后对对照组样品采用完全相同的实验条件,使用365nm波长紫外光交联,之后使用链霉亲和素修饰磁珠富集细胞表面膜蛋白。但与实验组不同的是,由于对照组没有膜靶向功能,在试剂孵育后的清洗步骤中加入0.05%tween 20,从而将探针从细胞中去除。因此在后续富集中所得产物中将不再包含细胞表面膜蛋白,只包含与磁珠非特异性吸附的蛋白。在此基础上将实验组与对照组样品使用胰蛋白酶酶解为肽段后
进行质谱分析。从而可以通过实验组和对照组中鉴定蛋白质种类和含量的差异获得更为准确的细胞表面膜蛋白鉴定结果,若对照组中未鉴定到或含量明显低于实验组的蛋白质则可认为是高可信的细胞表面膜蛋白。
[0068]
进一步,在上述方法中,c16-mbp试剂孵育时间为10-30min,优选为20min;365nm波长紫外交联功率为150w,照射时间为1-3min,优选为1min;孵育完成后,可通过磁性分离收集磁珠并弃去上清液,磁珠依次分别用200μl ripa(强)、200μl pbs溶液各清洗三次;通过磁分离弃去上清液,获得捕获了细胞表面膜蛋白的磁珠。后续加入2μl 0.5μg/μl的trypsin酶解蛋白质,进行蛋白质组分析。
[0069]
相对于现有技术,本发明具有的优点和有益效果:
[0070]
(1)本发明不仅为本领域提供了一种新的光标记试剂c16-mbp,而且还提供了一种基于光标记试剂c16-mbp化学标记的细胞膜表面蛋白及其糖基化的富集鉴定新方法,所述方法中采用的c16-mbp试剂含有一个c16基团,可以特异性的靶向细胞膜;一个聚乙二醇(peg)间隔剂,用于增加试剂的水溶性;两个4-(n-马来酰亚胺基)二苯甲酮(mbp)基团,用于与细胞表面蛋白的紫外交联;一个生物素基团,可以特异性地与商业化的链霉亲和素修饰磁珠结合,最终实现细胞表面膜蛋白的特异性富集。
[0071]
(2)与其他富集方法相比,本发明提供的基于c16-mbp化学标记的细胞表面膜蛋白富集鉴定新方法具有如下三大优势:
[0072]
①
相较于经典的利用超速离心的富集方法,基于c16-mbp试剂标记的方法能够快速对细胞表面膜蛋白进行标记和富集,节约了大量的时间和起始细胞量,在缩短了实验时间的同时也无偏性的对细胞膜表面蛋白进行标记和富集,该方法对细胞表面膜蛋白的富集鉴定、药物靶点的发现均具有重要意义;
[0073]
②
相较于酰肼化学、“点击化学”和基于细胞表面聚糖的酶标记方法,基于c16-mbp试剂标记的方法可以鉴定到更多的细胞表面蛋白,细胞表面靶向能力更好,其蛋白质组覆盖率和选择性较为理想。此外,聚糖的化学/酶标记可能会改变其组成和结构,从而导致ms难以完整地阐明聚糖/糖肽,多重富集过程中样品的显著损失可能也会严重降低鉴定规模,而利用faims技术可直接对富集到的膜蛋白进行分析,从而避免富集过程中的损失实现对糖肽的分离分析此外,c16-mbp试剂标记的效率更高,选择性和灵敏度更好;
[0074]
③
七肽肽链单元作为c16-mbp试剂中连接细胞膜靶向基团、光交联基团和生物素的连接臂,可有效增加探针的溶解度,同时由于其带正电性质使探针更容易靶向细胞膜,提高链霉素修饰磁珠对细胞表面蛋白的富集效率。
附图说明
[0075]
图1为本发明所述细胞表面膜蛋白富集方法的实验流程图;
[0076]
图2为本发明所合成的细胞膜定位探针c16-mbp的maldi-tof-ms表征图;
[0077]
图3为本发明所合成的细胞膜定位探针c16-mbp的膜定位效果荧光图;
[0078]
图4为本发明所述方法对细胞中细胞表面膜蛋白富集产物的sds-聚丙烯酰胺凝胶电泳图;其中,a图:对照组为实验过程中未加入c16-mbp探针、未进行365nm紫外照射或未进行链霉亲和素修饰磁珠富集处理,b图:对照组为不含c16探针与细胞孵育,以此验证c16-mbp探针对细胞表面膜蛋白富集的有效性;
[0079]
图5为采用本发明所述方法鉴定得到的结果图,其中,a图:本发明所述方法鉴定的ht22细胞中细胞表面膜蛋白的火山图(实验组与对照组蛋白质定量差异图);b图:对所鉴定符合卡值标准的细胞表面膜蛋白的go cellular component、go biological process和go molecular function分析,得到的前10个富集条目;
[0080]
图6为本发明所述方法四次技术重复组所得细胞表面膜蛋白的定量重现性评价图,其中,左下部分方框内的数字表示相应两组技术重复之间的pearson相关系数;
[0081]
图7为本发明所述方法四次技术重复组鉴定得到的结果图,其中,a图:本发明所述方法四次技术重复组所得细胞表面膜蛋白的定性重现性评价,b图:每次技术重复组所鉴定的总蛋白数和对应的细胞表面膜蛋白数;
[0082]
图8为本发明所述方法四次技术重复组鉴定得到的结果图,其中,a图:本发明所述方法四次技术重复组所得细胞表面膜糖蛋白go biological process和go molecular function分析,得到的前10个富集条目,b图:本发明所述方法四次技术重复组所得细胞表面膜糖蛋白和糖基化肽段的鉴定数目。
具体实施方式
[0083]
下面结合具体实施例,进一步阐述本发明,仅用于解释本发明,而不能理解为对本发明的限制。本领域的普通技术人员可以理解为:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
[0084]
实施例1新的光标记试剂c16-mbp的制备
[0085]
本实施例中制备得到的新的光标记试剂c16-mbp的结构式如式i所示。
[0086][0087]
1、首先合成用于制备新的光标记试剂c16-mbp的化合物2,即膜靶向控制探针c16-crrrrck-peg6-biotin(简称c16-pep-biotin)
[0088]
化合物2(c16-crrrrck-peg6-biotin)的具体合成步骤如下:
[0089]
(1)肽链都是从c端的氨基酸开始链接,向n端进行;
[0090]
(2)树脂活化:取2-cl树脂于洁净干燥的反应管中,加入适量dmf,活化30min左右;
[0091]
(3)氨基酸链接:称取计算得出量的c端首位氨基酸fmoc-asp(otbu)-nh2(被保护)
及diea 0.5ml加入到反应管,加入过量dmf作为溶剂反应,根据氨基酸不同加入不容类型催化剂;
[0092]
(4)洗脱fmoc保护;
[0093]
(5)检测:固相多肽合成中,主要是通过检测树脂上游离氨基来判断连接效率,检测方法为kaiser方法,其检测结果,如果有游离氨基的时候,显示蓝色,或红褐色(pro,ser,his),其中,kaiser试剂包括:a液(6%茚三酮的乙醇溶液)b液(80%苯酚的乙醇溶液),c液(2%0.001m kcn的吡啶溶液),取少量反应的树脂,加入a液、b液、c液各2-3滴,105-110℃下加热5min,如果溶液有蓝色,或树脂出现蓝色、红褐色,表明还有游离氨基,否则说明连接完全;
[0094]
(6)检测成功后继续链接c端第二个氨基酸。方法同上,从第(3)步起;
[0095]
(7)氨基酸接完后,加适量dmf,加入diea,然后加fitc反应4h,避光;
[0096]
(8)切割:三氟乙酸切割液切割2.5-5h,反应液抽滤,得多肽的三氟乙酸溶液;
[0097]
(9)沉淀:用过量乙醚沉淀,离心,离心样品用乙醚洗脱多次,离心,得初肽样品;
[0098]
(10)纯化:粗肽过hplc纯化,达到较高纯度;
[0099]
(11)质谱分析(检测);
[0100]
(12)冻干:液氮速冷,然后冻干备用。
[0101]
(13)质量分析(coa):高效液相色谱仪(hplc)和质谱仪(ms)对目标化合物进行肽序、分子量和化学纯度进行质量分析。
[0102]
(14)最终合成得到的化合物2(c16-crrrrck-peg6-biotin)的结构式如式ⅱ所示:
[0103][0104]
2、新的光标记试剂c16-mbp的合成
[0105]
所述新的光标记试剂c16-mbp由通过上述步骤合成得到的化合物2(c16-pep-biotin)与化合物3(4-(n-马来酰亚胺基)二苯甲酮)(来源sigma-aldrich,cas#:92944-71-3)发生迈克尔加成反应之后水解得到,其中,所述膜靶向控制探针c16-crrrrck-peg6-biotin(c16-pep-biotin,20mg,11.25μm)与4-(n-马来酰亚胺基)二苯甲酮(mbp,6.24mg,22.50μm)的投料摩尔比为1:2,所述迈克尔加成反应在水溶剂中进行,所述迈克尔加成反应的操作步骤为:先将化合物3溶解于水中,之后逐滴加入化合物2所示化合物,室温过夜反应后机械超声30min(20w)即可,具体操作步骤如下:
[0106]
将通过上述步骤合成得到的c16-crrrrck-peg6-biotin(c16-pep-biotin,20mg,11.25μm)和4-(n-马来酰亚胺基)二苯甲酮(mbp,6.24mg,22.50μm)分别溶解于1ml pbs(ph 7.4)中并混合均匀,在冰水浴中对上述混合样品进行超声波处理。采用脉冲超声(1s开,1s关)超声30分钟后,粗产物用脱盐柱进行纯化。收集纯化后的馏分,冷冻干燥。针对不含c16的非膜靶向控制探针,将crrrrck-peg6-biotin(pep-biotin,20mg,12.99μm)和4-(n-马来酰亚胺基)二苯甲酮(mbp,7.20mg,25.98μm)分别溶解于1ml pbs(ph 7.4)中并混合均匀,在
冰水浴中对上述混合样品进行超声波处理。采用脉冲超声(1s开,1s关)超声30分钟后,粗产物用脱盐柱进行纯化,作为后续实施例中的对照。采用基质辅助激光解吸电离飞行时间质谱(maldi-tof-ms)测定c16-pep-biotin和mbp-biotin探针的分子量。用流动相a(0.1%三氟乙酸-100%水)和b(0.1%三氟乙酸-100%乙腈),流速为1ml/min,采用液相色谱法测定两种探针的纯度。分析柱为shimadzu inertsil ods-sp,尺寸为4.6
×
250mm,粒度为5μm。检测波长为220nm。
[0107]
所述新的光标记试剂c16-mbp的具体反应式如下:
[0108][0109]
本发明所述细胞表面膜蛋白富集方法的实验流程图如图1所示;本发明制备得到的细胞膜定位探针(新的光标记试剂c16-mbp)的maldi-tof-ms表征图如图2所示,结果表明本发明成功制备得到包含膜靶向基团、七肽肽链基团、生物素基团、以及光交联基团的光标记试剂c16-mbp;本发明制备得到的细胞膜定位探针(新的光标记试剂c16-mbp)的膜定位效果荧光结果图如图3所示,结果表明了本发明制备得到的光标记试剂c16-mbp能够用于细胞表面膜蛋白的准确定位中。
[0110]
实施例2 c16-mbp试剂富集细胞中细胞表面膜蛋白的效果评价
[0111]
当15cm细胞培养皿中的ht22细胞(购自于上海泽叶生物科技有限公司)生长大约至80%覆板率时,除去培养皿中的培养基,使用冰pbs(4℃)清洗细胞3次(每次5ml)去除多余的培养基。加入c16-mbp试剂(终浓度为20μm),于4℃孵育20min,之后弃去溶液,pbs清洗3次,然后置于冰上,进行紫外光交联。紫外光波长为365nm,功率为150w,照射时间为1min。用细胞刮将细胞刮至1.5ml ep管中,1000g离心3min去除上清,然后加入ripa(强)使得总体积大约为400μl。冰上超声约5min(200w,2s on,2s off),16000g离心10min吸取上清至新的ep管中。加入50μl链霉亲和素固载磁珠(thermo),于4℃孵育1h。通过磁性分离弃去上清液,磁珠依次分别用200μl ripa(强)、200μl pbs溶液各清洗三次。通过磁分离弃去上清液,加入2μlβ-巯基乙醇和10μl loading buffer,95℃变性10min,离心取上清进行sds-聚丙烯酰胺凝胶电泳。
[0112]
如图4a所示,与实验组相比,未加入c16-mbp试剂的对照组蛋白条带明显较少,证明c16-mbp试剂在细胞表面膜蛋白的富集中起到关键作用。此外,实验过程中没有365nm紫外光照的对照组蛋白条带也相对较少,是因为没有365nm紫外交联探针无法与细胞表面蛋白共价结合,不会引入假阳性结果。
[0113]
如图4b所示,与实验组相比,相同实验条件下无c16的对照组探针蛋白条带明显减少。这是由于无c16的对照组探针没有膜靶向功能,在于细胞孵育后即可通过溶剂清洗干净,磁珠未捕获到蛋白,仅可能有少量非特异性吸附在磁珠上。以上实验证明了c16-mbp试剂对于细胞膜表面蛋白具有很好的选择性以及富集效果。
[0114]
实施例3 c16-mbp试剂富集细胞中细胞膜表面蛋白的蛋白质组质谱分析
[0115]
实验组:采用与实施例2中相同的实验条件,通过磁珠捕获获得纯化的细胞膜表面蛋白后,通过磁性分离弃去上清液,磁珠依次分别用200μl ripa(强)、200μl pbs溶液、200μl 50mm nh4hco3各清洗三次。通过磁分离弃去上清液后进行胰蛋白酶酶解,具体操作如下:加入10mm(终浓度)二硫苏糖醇56℃水浴中还原1小时,之后加入50mm(终浓度)碘乙酰胺于暗处放置30min进行烷基化处理,取变性后的蛋白加入1μg胰蛋白酶,放置于37℃水浴孵育12小时,通过磁性分离弃去磁珠后获取上清液及其中的酶解产物。酶解产物肽段经过c18 zip-tips脱盐小柱脱盐,洗脱液冷冻干燥后备用。
[0116]
对照组:取与实验组等量的细胞,其他操作与实验组完全相同。通过磁性分离弃去上清液,磁珠依次分别用200μl ripa(强)、200μl pbs溶液、200μl50mm nh4hco3各清洗三次。
[0117]
质谱分析:使用纳升液相色谱(easy-nlc 1200)与orbitrap exploris 480联用质谱仪对样本进行分析。其中,液相色谱使用20cm长的c18反向色谱填料装填柱(填料直径1.9μm,色谱柱内径75μm),并以300nl/min的流速来实现样品的分离。一级质谱分析的扫描范围设置为350-1500m/z,分辨率为60000。喷雾电压为2.2kv,离子传输管温度为320℃。agc为300%(3
×
106),最大注入时间为50ms,动态排除时间为45s。对于ms2,分辨率设定为15000,agc为75%(7.5e4),最大注入时间为22ms。选择前10个高含量的母离子(电荷为2-6)进行质谱分析。动态排除时间设置为30s。允许质量偏差为
±
10ppm,母离子强度阈值为2
×
104。对于hcd模式下的母离子破碎,使用了30%的碰撞能量。对于faims实验,使用cvs为-45和-60进行分析。
[0118]
质谱数据分析:质谱原始数据使用maxquant软件进行搜库解析。蛋白酶切模式设置为“胰蛋白/p”,并允许每个肽段中最多含有2个漏切位点(missed cleavage),每个肽段
最少含有6个氨基酸残基。脲甲基修饰半胱氨酸(carbamidomethyl cycteine,即被碘乙酰胺封闭的半胱氨酸)被设为固定修饰,甲硫氨酸的氧化和n端乙酰修饰被设为可变修饰。对于蛋白质鉴定,fdr上限设为1%。将p值小于0.01、且富集倍数大于等于4的蛋白质认为是高置信的细胞膜表面蛋白。
[0119]
如图5所示,本发明提供的方法鉴定出2835个高置信的细胞膜表面蛋白。对这些蛋白进行go功能分析,结果表明最富集的条目中绝大部分都是与细胞膜表面蛋白相关,进一步证明了该方法的可靠性和选择性。进一步对四次技术重复所得细胞膜表面蛋白进行相关性分析,结果如图6所示。各次技术重复之间的相关性可达0.9以上,证明该技术的实验重复性良好,可信度高。如图7所示,本发明所提供方法四次技术共鉴定到3103个细胞表面膜蛋白,重复鉴定到的细胞膜表面蛋白中有2560个(75.4%)至少被鉴定到两次,并且单次实验的选择性均大于65%,以上结果进一步证明该方法在细胞膜表面蛋白规模化富集方面的高选择性和广谱型。
[0120]
实施例4 c16-mbp试剂富集的细胞膜表面糖基化蛋白质组质谱分析
[0121]
考虑到富集的细胞膜表面蛋白样品量十分有限,串联n-糖肽富集可能不太实际。因此,在本实施例中,发明人提出了利用高场非对称波形离子迁移谱(faims)在不富集n-糖肽的胰蛋白酶消化肽中直接鉴定n-糖基化蛋白。c16-mbp探针富集的细胞表面蛋白被消化并直接进行ms分析,由于其分子量较大,不受非糖肽的抑制从而使完整的n-糖肽与之分离。
[0122]
如图8a显示,通过lc-ms分析,从c16-mbp探针的富集产物中鉴定出793个糖蛋白,包括1483条糖肽,表明该策略在大规模细胞表面n-糖蛋白组分析中的潜力。图8b对鉴定的n-糖蛋白进行分子功能和生物进程分析,“信号受体活性”(涉及信号转导、受体活性)和“分子运输活性”的分子功能,以及“细胞通讯”、“发育过程”和“信号转导”显著富集,这与报道的n-糖蛋白go功能一致。
[0123]
上述实施例的说明只是用于理解本发明的方法及其核心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也将落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1