一种提高葡萄糖氧化酶基因在毕赤酵母中表达效率的方法
1.本发明涉及基因工程技术领域,具体涉及一种葡萄糖氧化酶基因在毕赤酵母中的高效表达方法。
背景技术:
2.葡萄糖氧化酶(glucose oxidase,god)的系统命名为β-d-葡萄糖氧化还原酶(ec1.1.3.4)。最早于1928年由muller在黑曲霉(aspergillus niger)中发现,god能够以氧分子为电子受体,催化β-d-葡萄糖生成葡萄糖酸和过氧化氢。另外,god对甘露糖、半乳糖和木糖也具有较为缓慢的氧化作用。该酶已被广泛地应用于食品工业和医学诊断和研究中。葡萄糖氧化酶是一种重要的新型饲料用酶制剂,能够保障畜禽肠道健康,提高消化性能,促进动物生长,并能够在一定程度上缓解霉菌毒素中毒症状。
3.god是一种同型二聚体酶,两个亚基通过两个二硫键结合,每一个亚基都含有两个不同的区域:一个区域以非共价键的方式与黄素腺嘌呤二核苷酸(flavin adenine dinucleotide,fad)结合,该区域主要为β-折叠;另一个区域结合底物β-d-葡萄糖,该区域由四个α-螺旋支撑一个反向平行的β-折叠。研究表明,相比于单体,二聚体形式的稳定性更强;当fad从单体酶上解离时,单体的稳定性变得更差,这表明fad在god活性或稳定性方面均有重要的作用。
4.葡萄糖氧化酶氧化β-d-葡萄糖的过程分为分为两个半反应,如图1所示,在还原半反应中,葡萄糖氧化酶氧化β-d-葡萄糖生成d-葡萄糖-δ-内酯,d-葡萄糖-δ-内酯自发降解为葡萄糖酸,随后葡萄糖氧化酶上的辅基fad被还原为fadh2;在氧化半反应中,fadh2被o2氧化变成fad并生成h2o2。细胞内fad的合成在fad合成酶(fads)的催化下进行,因此,通过fad合成酶的过表达,有可能提高fad的合成量,进而提高葡萄糖氧化酶的表达量。
技术实现要素:
5.本发明的目的是提供一种高表达具有稳定活性的葡萄糖氧化酶的方法。
6.第一方面,本发明提供一种优化的葡萄糖氧化酶基因序列,本发明用毕赤酵母高频率密码子替换低频率密码子,并剔除ecori、xba i和sac i等酶切位点。并且,本发明还通过人工手动调整,调整序列中a、t碱基的含量,避免存在多个连续的a、t碱基,获得了能够与fad合成酶在同一重组菌中共表达的葡萄糖氧化酶基因序列。
7.具体地,本发明提供葡萄糖氧化酶基因序列如seq id no.2所示。
8.第二方面,本发明请求保护含有如seq id no.2所示葡萄糖氧化酶基因序列的表达载体。
9.第三方面,本发明请求保护含有如seq id no.2所示葡萄糖氧化酶基因序列的重组菌。
10.本发明所提供的重组菌中同时表达seq id no.2所示的葡萄糖氧化酶基因序列和seq id no.3所示的黄素腺嘌呤二核苷酸合成酶基因序列。
11.本发明还提供一种提高葡萄糖氧化酶活性及表达效率的方法,构建seq id no.2所示的葡萄糖氧化酶基因序列和seq id no.3所示的黄素腺嘌呤二核苷酸合成酶基因序列共表达的重组菌。
12.在本发明提供的方法中,采用载体ppiczαa构建含有seq id no.2所示的葡萄糖氧化酶基因序列的重组表达载体ppic-god-opt;采用载体pgapka构建含有seq id no.3所示的黄素腺嘌呤二核苷酸合成酶基因序列的表达载体pgap-fad。
13.具体地,本发明提供的方法,包括:
14.(1)对seq id no.1所示基因序列进行优化,获得seq id no.2所示葡萄糖氧化酶基因序列god-opt,在god-opt的5’和3’末端引入ecori和xba i酶切位点;
15.(2)将god-opt与ppiczαa载体分别用ecori和xba i酶切,获得重组表达载体ppic-god-opt;
16.(3)ppic-god-opt用sac i限制性内切酶线性化,将线性化的ppic-god-opt转入毕赤酵母感受态细胞中,获得重组菌x33/god;
17.(4)seq id no.3所示的基因序列与表达载体pgapka双酶切获得重组表达载体pgap-fad;
18.(5)将重组表达载体pgap-fad转入重组菌x33/god中,获得fad合成酶基因与god基因共表达菌株x33/god-fad。
19.在本发明提供的方法中,合成seq id no.3所示的基因序列所用引物序列如seq id no.6-7所示。
20.根据本领域技术人员的理解,本发明请求保护如seq id no.2所示的葡萄糖氧化酶基因序列或上述的重组菌在提高葡萄糖氧化酶表达量中的应用。
21.以及,如seq id no.2所示的葡萄糖氧化酶基因序列或上述的重组菌在提高葡萄糖氧化酶表达活性中的应用。
22.本发明的有益效果在于,通过fad合成酶的共表达,提高了god的表达效率。
23.本发明通过对毕赤酵母高频率密码子替换低频率密码子,并剔除ecori、xba i和sac i等酶切位点,并进行人工手动调整,调整序列中a、t碱基的含量,获得了能够与fad合成酶在同一重组菌中共表达的葡萄糖氧化酶基因序列如seq id no.2所示。
24.本发明将seq id no.2所示的基因序列与seq id no.3所示黄素腺嘌呤二核苷酸(fad)合成酶基因序列同时构建在重组菌中;fad合成酶基因与god基因共表达菌株产生的葡萄糖氧化酶发酵活力达到god基因单表达菌株的2.2倍;并且fad合成酶基因与god基因共表达菌株产生的葡萄糖氧化酶表达量得到显著提升。
附图说明
25.图1为本发明fad合成酶基因pcr扩增结果。其中,泳道1为dna marker;泳道2为fad合成酶基因。
26.图2为摇瓶发酵god的相对催化活力。
27.图3为发酵上清液sds-page电泳图。其中,m为蛋白分子量标准;泳道1为x33/god发酵上清液;泳道2为x33/god-fad-1发酵上清液;泳道3为x33/god-fad-2发酵上清液;泳道4为x33/god-fad-3发酵上清液;泳道5为x33/god-fad-4发酵上清液。
具体实施方式
28.以下实施例用于说明本发明,但不用来限制本发明的范围。
29.本发明实施例中缩影巴斯德毕赤酵母表达载体ppiczαa及菌株x33购自invitrogen公司。pgapka表达载体由本实验室构建并保存。大肠杆菌top10感受态细胞购自北京擎科新业生物技术有限公司。
30.本发明实施例中所用bca蛋白定量分析试剂盒,生产厂家为美国pierce公司产品;pr1910蛋白分子量标准和辣根过氧化物酶,生产厂家为中国索莱宝公司产品;抗生素zeocin,生产厂家为美国invitrogen;葡萄糖氧化酶标准品、邻联茴香胺,生产厂家为美国sigma-aldrich;限制性内切酶、phusion dna聚合酶,生产厂家为美国neb公司;g418,生产厂家为美国gibco公司。
31.实施例1黑曲霉god基因优化合成与表达菌株构建
32.(1)序列优化设计与表达载体构建
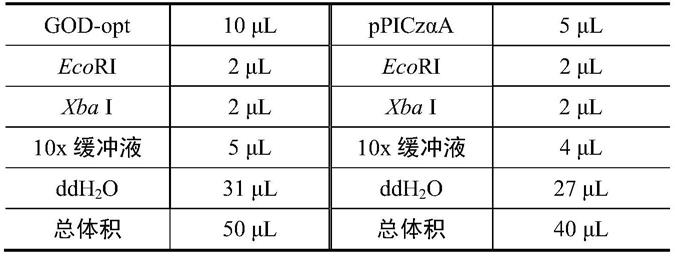
33.对genbank公开的黑曲霉god基因序列seq id no.1(登录号:mh593586.1)进行优化设计,用毕赤酵母高频率密码子替换低频率密码子,并剔除ecori、xba i和sac i等酶切位点,手动调整序列中at碱基的含量,避免存在多个连续的a、t碱基。然后进行全基因合成(序列见seq id no.2)。合成序列命名为god-opt,在god-opt的5’和3’末端分别引入了ecori和xba i酶切位点。god-opt与ppiczαa载体分别用ecori和xba i酶切,具体反应体系如表1所示。
34.表1 god-opt和ppiczaa的酶切体系
[0035][0036]
37℃保温5h。然后用dna柱纯化试剂盒(omega公司产品)纯化,再进行连接反应,反应体系如表2所示。
[0037]
表2连接反应体系
[0038][0039]
16℃保温12h,转化大肠杆菌top10感受态细胞(购自北京擎科新业有限责任公司),涂布含抗生素zeocin的低盐lb平板(10g/l胰蛋白胨、5g/l酵母提取物、5g/l nacl、
15g/l琼脂,25mg/l zeocin)。37℃培养过夜,挑取单菌落在低盐lb平板上划线培养。提取质粒,用引物aox-f和aox-r测序,确认序列连接正确。重组表达载体命名为ppic-god-opt。引物序列如表3所示。
[0040]
表3测序所用引物序列
[0041][0042]
(2)重组菌株构建:
[0043]
ppic-god-opt用sac i限制性内切酶线性化,37℃酶切12h以上,然后用dna柱纯化试剂盒纯化酶切产物,溶于10μl灭菌ddh2o中。具体反应体系见表4所示。
[0044]
表4重组表达载体线性化
[0045][0046]
挑毕赤酵母x33单菌落接种5ml ypd培养基(1%酵母提取物、2%蛋白胨、2%葡萄糖),30℃220rpm振荡培养过夜。按1%比例转接50mlypd培养基,30℃220rpm振荡直至od
600
约为1.0左右,4℃5000rpm离心5min收集菌体,用冰冷的灭菌水洗涤2-3次,菌体溶于1ml 1m山梨醇中,即为感受态细胞。将10μl线性化的ppic-god-opt加入100μl感受态细胞中,混匀,加入电击杯中,2000v电击5ms,立即加入1ml冰冷的1m山梨醇溶液,然后在30℃恢复培养3h。涂布含100μg/ml zeocin的ypds平板(1%酵母提取物、2%蛋白胨、2%葡萄糖、1.5%琼脂、1m山梨醇),30℃培养3~4天,待菌落清晰为止。重组菌株命名为x33/god。
[0047]
(3)重组菌株鉴定
[0048]
接种重组菌株x33/god于5ml ypd培养基,30℃220rpm振荡培养过夜。各取100μl接种于10ml bmgy培养基,继续30℃220rpm振荡培养24h。4℃5000rpm离心5min收集菌体,用真菌dna提取试剂盒(omega公司产品)提取基因组dna。进行pcr扩增。扩增体系如表5所示。扩增条件:94℃30s,55℃20s,72℃2min,30个循环。琼脂糖凝胶电泳检测扩增产物。
[0049]
表5 pcr扩增体系
[0050][0051]
实施例2毕赤酵母fad合成酶基因克隆与表达载体构建
[0052]
(1)fad合成酶基因克隆:
[0053]
根据genbank公开的毕赤酵母基因组序列(登录号:lt962476)设计引物,扩增其fad合成酶序列(seq id no.3),引物序列如表6所示。接种毕赤酵母x33于ypd液体培养基中,30℃振荡培养18h,5,000rpm离心5min收集菌体。用真菌dna提取试剂盒(omega公司产品)提取基因组dna。
[0054]
表6克隆fad合成酶基因所用引物序列
[0055][0056]
pcr扩增fad合成酶基因。扩增体系见表7。扩增条件:94℃30s,58℃20s,72℃1min,30个循环。琼脂糖凝胶电泳检测扩增产物。扩增出1条约800bp的条带。
[0057]
表7 fad合成酶基因pcr扩增体系
[0058][0059][0060]
(2)表达载体构建:
[0061]
pcr产物与表达载体pgapka双酶切反应体系如表8所示。
[0062]
表8 fad合成酶基因与表达载体pgapka的双酶切
[0063][0064]
37℃保温5h。然后用dna柱纯化试剂盒(omega公司产品)纯化,再进行连接反应,反应体系如表9所示。
[0065]
表9连接反应体系
[0066][0067]
16℃连接12h。转化大肠杆菌top10感受态细胞。涂布含抗生素zeocin的低盐lb平板。37℃培养过夜,挑取单菌落在低盐lb平板上划线培养。提取质粒,用引物aox-r测序,确认序列连接正确。重组表达载体命名为pgap-fad。
[0068]
实施例3fad合成酶基因与god基因共表达菌株构建
[0069]
挑x33/god单菌落接种5ml ypd培养基,30℃220rpm振荡培养过夜。按1%比例转接50ml ypd培养基,30℃220rpm振荡直至od
600
约为1.0左右,4℃5,000rpm离心5min收集菌体,用冰冷的灭菌水洗涤2-3次,菌体溶于1ml 1m山梨醇中,即为感受态细胞。
[0070]
重组质粒pgap-fad用限制性内切酶bgl ii线性化,酶切体系如表10所示。37℃保温5h。然后用dna柱纯化试剂盒(omega公司产品)纯化,dna溶解于10μl ddh2o。
[0071]
表10 pgap-fad的线性化
[0072][0073]
将10μl线性化的pgap-fad电击转化x33/god感受态细胞,电击条件为2000v、5ms,涂布含100μg/ml抗生素g418的ypds平板(1%酵母提取物、2%蛋白胨、2%葡萄糖、1.5%琼脂、1m山梨醇),30℃培养3-4天,待菌落清晰为止。重组菌株命名为x33/god-fad。
[0074]
实施例4摇瓶发酵
[0075]
(1)摇瓶发酵培养:
[0076]
挑取重组毕赤酵母菌株x33/god、x33/god-fad-1、x33/god-fad-2、x33/god-fad-3和x33/god-fad-4(均为随机挑选的克隆)分别接种ypd液体培养基,30℃220rpm振荡培养过夜。1%比例转接于20ml bmgy(1%酵母提取物,2%蛋白胨,1.34%ynb,4
×
10-5%biotin,1%甘油,100mm ph 6.0磷酸盐缓冲液)培养基的250ml摇瓶中,于30℃、220rpm摇床培养至od
600
值为2~6,5000rpm、4℃离心5min收集菌体。菌体用20ml bmmy培养基(1%酵母提取物,2%蛋白胨,0.1mol/l磷酸缓冲液ph 6.0,1.34%ynb,4
×
10-5%biotin)重悬菌体,于30℃、220rpm摇床培养,并加入终浓度为0.5%(v/v)的无水甲醇进行摇瓶诱导培养,每24h添加一次甲醇,离心取上清,4℃保存待测。
[0077]
(2)酶活分析:
[0078]
将葡萄糖氧化酶标准品稀释成0.4、0.8、1.2、1.6、2.0、2.4u/ml,在1.5ml离心管中加入500μl邻联茴香胺缓冲液,60μl 1m葡萄糖和20μl的100u/ml辣根过氧化物酶溶液,在测定温度下保温5min,加入20μl稀释好的葡萄糖氧化酶标准品,反应3min后加入100μl的2mol/l硫酸终止反应,取200μl反应液加入96孔板,使用酶标仪测定在460nm下反应液吸光值,做酶活-吸光值曲线,即为该反应条件下的标准曲线。
[0079]
将粗酶液稀释相应倍数,同上步骤,替代标准品,测定其吸光值,以先加反应终止液再加酶液作为空白对照,根据标曲计算该反应条件下的酶活。结果如图2所示,以菌株x33/god的发酵活力为100%,x33/god-fad-1的发酵活力达到了220%,x33/god-fad-2的发酵活力达到228%,x33/god-fad-3的发酵活力达到235%,x33/god-fad-4的发酵活力达到216%。说明fad合成酶的转入,显著提高了god的发酵活力。
[0080]
(3)上清液蛋白sds-page电泳:
[0081]
制备12.5%的sds-page胶,发酵上清液各取10μl上样,比较蛋白条带的亮度。如图3所示,共表达fad合成酶的蛋白条带亮度明显高于x33/god,说明fad合成酶的转入,提高了god的蛋白表达量。用bsa定量试剂盒对所表达蛋白定量分析,共表达fad菌株的god表达量提高了约1倍(见表11)。
[0082]
表11重组菌株发酵上清液蛋白定量和发酵活力结果
[0083]
菌株编号蛋白含量(mg/ml)发酵活力(u/ml)x33/god1.32145x33/god-fad-12.90320x33/god-fad-23.01330x33/god-fad-33.10340x33/god-fad-42.85313
[0084]
对比例1
[0085]
本对比例提供仅通过软件优化,未经手动调整的god基因序列序列如seq id no.8所示。
[0086]
本对比例获得seq id no.8的过程,与实施例1的序列优化方法相同,区别在于,本对比例所用的如seq id no.8的god基因序列完全由软件生成,没有经过人为调整。
[0087]
采用seq id no.8所示基因序列,构建共表达菌株时,发现fad合成酶基因与god基因共表达菌株产生的葡萄糖氧化酶发酵活力为140u/ml,与god基因单表达菌株的发酵活力,基本类似。并且本对比例得到的葡萄糖氧化酶发酵活力显著低于实施例3得到的共表达
菌株的god发酵活力。
[0088]
采用seq id no.8所示基因序列,构建共表达菌株时,发现fad合成酶基因与god基因共表达菌株的god表达量为1.30mg/ml,与god基因单表达菌株的god表达量,基本类似。
[0089]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1