一种提高人表皮生长因子分泌生产效率的方法
1.本发明属于基因工程技术和发酵工程领域,具体涉及到一种利用大肠杆菌提高人表皮生长因子分泌生产效率的方法。
背景技术:
2.生物合成医药蛋白越来越受到人们的广泛关注,因为与化学合成相比,生物合成的安全性更高。大肠杆菌具有很多优势,如生长周期短、易于实现高密度培养、遗传背景明确以及外源基因转化速度快等,因此常用于表达重组蛋白。然而,实现重组蛋白的商业化生产仍然是一个挑战,最重要的问题之一是大肠杆菌分泌蛋白质的能力很差。在生产过程中包涵体的形成、蛋白降解、细胞代谢负担或表达蛋白的转运/运输系统效率低下等问题都会导致蛋白分泌效率低下。为了提高大肠杆菌的蛋白质分泌效率,人们尝试了很多方法,如选择高效的信号肽(如ompa和pelb的信号序列)将蛋白质有引导至大肠杆菌蛋白分泌通路;对参与跨膜运输的关键转运蛋白或辅助蛋白进行修饰也被应用于提高蛋白质的分泌效率;此外,融合蛋白和分子伴侣的共表达也被证明可以增加蛋白质的表达和胞外分泌;其他改善蛋白质分泌的方法还包括优化发酵过程参数和培养基配方。
3.重组人表皮生长因子(rhegf)是一种潜在的治疗性蛋白,被广泛用作各种慢性伤口的愈合剂,已成功地在大肠杆菌bl21(de3)中使用pelb信号肽进行细胞外表达(中国专利号cn111471636a)。将hegf分泌到培养基中大大简化了产品的回收,并减少了下游发酵过程中的分离成本。然而,hegf的分泌效率不高且通常是以传统的游离发酵方式进行生产。在剪切力等压力条件下产人表皮生长因子细胞活力下降,不能连续化使用。目前已有一些提高人表皮生长因子蛋白分泌的研究,主要是通过优化密码子(中国专利号cn1360022a)、优化信号肽(中国专利号cn1854294a)、温度诱导(中国专利号cn102952817a)等方式实现产量提高。
4.为了提高hegf蛋白分泌效率,本研究采取不同的思路以开发新的策略,如尝试从大肠杆菌生物被膜等生理过程相关的角度,考察了生物被膜形成相关基因或一些其他未知基因的表达对人表皮生长因子分泌效率的影响,并进一步创建基于生物被膜的连续发酵过程。大肠杆菌生物被膜由嵌入胞外聚合物(eps)基质中的菌落组成,是一种复杂的细胞群落。包裹在生物膜中的细胞吸附在固体介质表面,能够通过吸收营养不断地进行自我更新并承受不利的条件。基于生物被膜的固定化连续发酵体系已应用于l-苏氨酸等小分子产品(中国专利号cn201910392765.7)的生产中,但是目前还没有应用于人表皮生产因子的报道,且大肠杆菌生物被膜相关基因(如bcsb、fimh、csgab)以及其他一些生理基因(如moae、gshb、ycea、ychj)对hegf分泌和生产的影响也未见报道。
技术实现要素:
5.发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种重组大肠杆菌及其构建方法。
6.本发明还要解决的技术问题是提供一种提高人表皮生长因子分泌生产效率的方法。
7.为了解决上述第一个技术问题,本发明公开了一种重组大肠杆菌,对大肠杆菌采用以下任意一种或几种方式进行改造;
8.a.弱化moae基因、gshb基因、ycea基因、ychj基因中的一个或几个基因的表达;
9.b.强化bcsb基因、csga基因、csgb基因、fimh基因中的一个或几个基因的表达。
10.其中,所述moae基因(ncbi gene id:945399)编码一个参与钼蝶呤生物合成的蛋白质;gshb基因(ncbi gene id:947445)编码一种谷胱甘肽合成酶;ycea(ncbi gene id:945601)基因编码一种trna u34羟化酶;ychj基因(ncbi gene id:945828)编码一种ntf2类似结构域蛋白。
11.其中,所述bcsb基因(gene id:948045)参与纤维素合成,csga基因(gene id:949055)、csgb基因(gene id:947391)参与卷曲菌毛的合成,fimh基因(gene id:948847)参与i型菌毛的合成。
12.其中,所述弱化基因的表达方式为在大肠杆菌基因组中进行基因敲除;所述基因敲除方式为crispr/cas9编辑技术(jiang,y,et al.,multigene editing in the escherichia coli genome via the crispr-cas9 system.appl environ microbiol.2015jan 30.81(7),2506-14.)、或λ-red同源重组技术(madyagol,m.et al.,2011.gene replacement techniques for escherichia coli genome modification.folia microbiol(praha),56(3),253-63.)。
13.其中,所述强化基因表达的方式为在大肠杆菌基因组中进行整合表达,整合的方式采用上述crispr/cas9编辑技术或同源重组技术;或将目的基因连接到基因表达质粒上,如pbbe1a、pet28、petduet等常见基因表达质粒进行表达。
14.其中,通过基因组整合方式表达bcsb基因、csgacsgb基因和fimh基因的大肠杆菌重组菌株命名为bl21-bcsb*、bl21-csgacsgb*和bl21-fimh*;通过质粒表达bcsb基因、csgacsgb基因和fimh基因的大肠杆菌重组菌株命名为bl21-bcsb
+
,bl21-csgacsgb
+
,bl21-fimh
+
。
15.其中,所述bl21-bcsb*、bl21-csgacsgb*、bl21-fimh*的基因整合位点可以选择已有文献报道的大肠杆菌基因整合位点,比如yjip_yjir、thrw_ykfn、ykgh_beta、iley_ygaq(goormans,a.r.et al.,2020.comprehensive study on escherichia coli genomic expression:does position really matter?metab eng,62,10-19.),也可以选择上述moae基因、gshb基因、ycea基因、ychj基因等敲除位点的任一个,整合片段为启动子+目的基因片段+终止子构成的表达盒;优选地,整合位点为moae敲除位点,表达盒中启动子为trc启动子,所述基因组整合方式为crispr/cas9编辑技术或λ-red同源重组技术
16.上述重组大肠杆菌的构建方法也在本发明的保护范围之内。
17.为了解决上述第二个技术问题,本发明公开了一种提高人表皮生长因子分泌生产效率的方法,将上述重组大肠杆菌于培养基中进行发酵,待菌体生长的光密度(od
600
)数值达到0.6~0.8之间,使用iptg诱导后进行人表皮生长因子的生产,即得含人表皮生长因子的发酵液;其利用上述重组大肠杆菌,通过导入产人表皮生长因子的基因,在培养基中进行发酵,可以将人表皮生长因子更加有效地分泌到液体培养基中进行生产,同时也可以建立
起高效的连续化发酵体系。
18.其中,所述培养基含有胰蛋白胨5~15g/l,酵母粉提取物1~30g/l,氯化钠0~12g/l,葡萄糖0~50g/l,甘油0~10ml/l,磷酸氢二钾0~14.4g/l,磷酸二氢钾0~5g/l。
19.其中,所述培养基中,当氯化钠含量不为0g/l时,甘油、磷酸氢二钾和磷酸二氢钾的含量均为0g/l,即所述培养基含有胰蛋白胨5~15g/l,酵母粉提取物1~30g/l,氯化钠0.001~12g/l,葡萄糖0~50g/l,优选为胰蛋白胨8~12g/l,酵母粉提取物2~7g/l,氯化钠8~12g/l,葡萄糖0~6g/l,进一步优选为胰蛋白胨10g/l,酵母粉提取物5g/l,氯化钠10g/l,葡萄糖0或5g/l。
20.其中,所述培养基中,当氯化钠含量为0g/l时,葡萄糖的含量为0g/l,甘油、磷酸氢二钾和磷酸二氢钾的含量均不为0g/l,即所述培养基含有胰蛋白胨5~15g/l,酵母粉提取物1~30g/l,甘油0.001~10ml/l,磷酸氢二钾0.001~14.4g/l,磷酸二氢钾0.001~5g/l。
21.其中,所述发酵过程中加入异丙基-β-d-硫代半乳糖苷iptg;优选地,所述发酵过程中加入0.01~1.5mm异丙基-β-d-硫代半乳糖苷;优选地,所述发酵过程中加入1mm异丙基-β-d-硫代半乳糖苷。
22.其中,所述发酵的温度为25~37℃;优选地,所述发酵的温度为25℃。
23.其中,所述发酵为游离发酵或固定化发酵;所述发酵为单批次发酵、连续发酵或反复批次发酵;所述发酵为固定化反复批次发酵。
24.其中,所述固定化发酵的载体为棉纤维、聚酯纤维、活性炭、无纺布、聚乳酸、尼龙纤维、木浆棉、活性炭、聚乙烯、聚乙烯醇、丝绸、聚氨酯、粘土和金属中的任意一种或多种组合。其中,所述载体以所述材质本身存在,或以其加工所得布条、树脂、海绵、海绵类似物、塑料片、塑料弹簧和载玻片中的任意一种形态存在。
25.其中,所述固定化发酵中载体的用量为5~100g/l;优选地,所述固定化发酵中载体的用量为5~60g/l;进一步优选地,所述固定化发酵中载体的用量为10~50g/l。
26.本发明中,所述游离发酵,是指在培养基中,不加入固体载体,细胞以游离悬浮的状态存在于培养基中进行发酵。
27.本发明中,所述固定化发酵,是指在培养基中加入固体载体,细胞在固体载体上吸附生长,进行发酵。
28.本发明中,所述单批次发酵是指在培养基中,接种后不再向发酵液加入或移出任何营养物质直至发酵结束(18~72h)。
29.本发明中,所述连续发酵是指在发酵过程中放出一定量的发酵液,同时向反应器内添加新鲜的培养基,从而实现发酵过程的连续化。放出发酵液的方式可以是间歇式的,比如每次可放出反应体积的4/5、2/3、1/2、1/3或1/4等等,然后再补充同等体积的新鲜培养基进行发酵;放出发酵液的方式也可以是连续式的,即连续地流出一部分发酵液,同时连续地补充同等体积的发酵液。
30.本发明中,所述固定化反复批次发酵,其作为连续发酵的特殊方式之一,是指在培养基中加入固体载体,当发酵过程中一个周期结束后,将发酵液全部放出,保留固体载体,然后加入新的发酵培养基继续进行下一批次的发酵,如此反复进行。
31.有益效果:与现有技术相比,本发明具有如下优势:
32.1、通过moae基因、gshb基因、ycea基因、ychj基因的弱化或者bcsb基因、csgacsgb
基因、fimh基因表达的强化,能够有效提高大肠杆菌细胞在多种发酵条件下分泌人表皮生长因的效率,并有利于细胞在多种固体表面吸附生长形成生物被膜。
33.2、本发明了一种大肠杆菌固定化发酵产人表皮生长因子的方法,利用固体材料作为大肠杆菌吸附生长的固定化载体,吸附固定的菌体可以反复利用,提高菌体密度,无需在发酵过程中再制备种子,能够进行长期连续发酵,从而提高了发酵速度,减少了停工时间,提高了生产强度,节约了培养资源和发酵成本。
34.3、本发明提供了一种通过生物被膜提高人表皮生长因子生产效率的方法,提高了大肠杆菌分泌生产人表皮生长因子的能力,使得大肠杆菌在单批次游离发酵和固定化连续发酵中产人表皮生长因子的效率得到了1~2倍的提高,实现了人表皮生长因子的连续化生产。
附图说明
35.下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
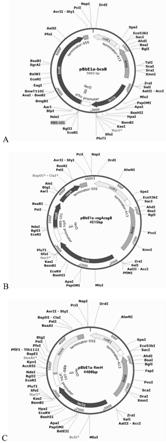
36.图1为过表达bcsb、csgacsgb、fimh的质粒图谱。
37.图2为将目的基因连接到质粒pbbe1a热转至bl21后挑取阳性转化子提质粒进行酶切验证结果,从左至右边依次为pbbe1a-bcsb酶切结果、pbbe1a质粒酶切结果、pbbe1a-csgacsgb酶切结果、pbbe1a质粒酶切结果、pbbe1a-fimh酶切结果、pbbe1a质粒酶切结果。
38.图3依次为bl21中敲除moae、gshb、ycea、ychj基因挑取阳性转化子菌落pcr验证的核酸电泳图(以野生菌bl21菌落pcr结果作为对照)。
39.图4依次为bl21中moae敲除位点整合bcsb、csgacsgb、fimh基因挑取阳性转化子菌落pcr验证的核酸电泳图(以野生菌bl21菌落pcr结果作为对照)。
40.图5为96孔板结晶紫染色相对生物量,a图中对照为在野生bl21中导入产人表皮生长因子的质粒pet30a-hegf以及空质粒pbbe1a;b图中对照只包含一个质粒pet30a-hegf。
41.图6为使用lb培养基,iptg浓度为1mm时单批次发酵sds-page凝胶电泳结果及使用image lab对蛋白胶进行相对定量分析的结果。
42.图7为使用lb培养基,以棉纤维为固定化载体时固定化连续发酵发酵液中hegf sds-page凝胶电泳结果;图7a中泳道条带依次为bsa,对照1,bl21-bcsb
+
1,bl21-csgacsgb
+
1,bl21-fimh
+
1,对照2,bl21-bcsb
+
2,bl21-csgacsgb
+
2,bl21-fimh
+
2(对照1和对比2是平行试验,均为在bl21(de3)中导入空质粒pbbe1a和产人表皮生长因子的质粒);图7b中泳道条带依次为bsa,对照(在b21(de3)中导入产人表皮生长因子的质粒),bl21δmoae,bl21δgshb,bl21δycea,bl21δmoae,bl21-bcsb*,bl21-csgacsgb*,bl21-fimh*。
43.图8为使用lb培养基,以棉纤维为固定化载体时固定化连续发酵使用image lab软件对hegf进行相对定量分析的结果。
44.图9为使用lb培养基,以棉纤维为固定化载体时固定化连续发酵发酵液中的菌体光密度od
600
。
具体实施方式
45.根据下述实施例,本领域的技术人员更易理解本发明内涵。实施例所描述的内容
仅用于说明本发明,不应当也不会限制权利要求书中所详细描述的本发明,而利用菌株固定化表达重组蛋白也将包含于本发明范围。
46.在本发明所述的技术方案为前提进行实施,实施例给出了详细的实施方式和具体的操作过程。下属实施例中所用试剂均可从商业途径获得。下属实施例中所述实验方法,如无特殊说明,均为常规方法。
47.下列实施例以人表皮生长因子为目标产物,通过基因改造以及表面固定化连续化发酵,详细阐明本发明方法。
48.下列实施例中所使用的大肠杆菌为bl21(de3)。
49.以下实施例中所使用的酵母粉提取物为oxoid。
50.以下实施例中所涉及的重组菌bl21-bcsb
+
,bl21-csgacsgb
+
,bl21-fimh
+
,bl21-bcsb*,bl21-csgacsgb*,bl21-fimh*,bl2δmoae,bl21δgshb,bl21δycea和bl21δychj分别是以大肠杆菌bl21(de3)为起始菌。
51.以下实施例中所述对照菌大肠杆菌bl21(de3),若无特殊说明,所述对照菌大肠杆菌bl21(de3),若是与通过质粒pbbe1a表达目的基因的重组大肠杆菌相比,所述对照菌大肠杆菌bl21(de3)在野生bl21(de3)中导入产人表皮生长因子的质粒pet30a-hegf以及空质粒pbbe1a;若是与采用另外两种方式改造(整合表达和基因敲除)的重组大肠杆菌相比,所述对照菌大肠杆菌bl21(de3)只导入了产人表皮生长因子的质粒pet30a-hegf。
52.产人表皮生长因子的质粒是以seq id no.1为人表皮生长因子基因,使用质粒pet30a按照中国发明申请:cn 111471636 a构建得到的,命名为pet30a-hegf。
53.将产人表皮生长因子的质粒导入构建成功的底盘菌株中,得到卡那抗性的单质粒菌株(对大肠杆菌基因组进行改造)和卡那+氨苄抗性的双质粒菌株(通过质粒pbbe1a表达目的基因)。
54.菌株构建过程中使用软件snap gene进行引物设计,相关引物序列seq id no.2-65等合成及测序由通用公司合成提供。
55.实施例中的分子生物学实验包括质粒构建、酶切、感受态细胞制备、转化等按照《分子克隆实验指南》进行。
56.实施例1构建通过质粒过表达bcsb,csgacsgb,fimh基因的重组菌bl21-bcsb
+
,bl21-csgacsgb
+
,bl21-fimh
+
57.1.1按照质粒提取试剂盒(axygen axyprep plasmid miniprep kit)的步骤提取质粒pbbe1a,选取bglii位点和avrii位点将载体进行双酶切。
58.1.2以大肠杆菌bl21基因组为模板进行目的片段的pcr,以质粒pbbe1a为模板,对terminator部分进行pcr后,将两个片段通过overlap pcr连接起来用于后续质粒构建。使用snapgene软件设计目的基因片段引物,核苷酸序列见seq id no.2-11(质粒pbbe1a上的选择的两个酶切位点中bglii位点在trc启动子之后,而terminator在avrii位点之前,seq id no.4、5是作为上下游引物以这个质粒为模板将terminator进行pcr后,与bcsb基因overlap连接起来后再通过一步克隆连接到质粒pbbe1a上。seq id no.5可同时作为三个基因overlap pcr的下游引物;seq id no.8和seq id no.11分别为csgacsgb和fimh基因terminator的上游引物)
59.pcr体系(100μl分装为5管):5
×
ps buffer 20μl,dntp mix 10μl,上、下游引物各
1μl,模板1μl,prime star酶1μl,dd h2o 67μl
60.pcr条件:95℃,10min;95℃,10s;55℃,15s;72℃,2min(延伸时间根据片段长度而变化),38个循环。
61.1.3重组质粒的构建
62.将步骤1.1得到的纯化线性化载体与步骤1.2得到的目的基因片段按照clonexpress ii one step cloning kit试剂盒的说明书的步骤相连接,得到重组质粒pbbe1a-bcsb,pbbe1a-csgacsgb,pbbe1a-fimh,过表达bcsb、csgacsgb、fimh的质粒图谱如图1所示。
63.1.4将构建好的重组质粒热转至bl21感受态中,在含有100μg/ml氨苄抗性的lb平板上筛选阳性转化子。
64.1.5挑取阳性转化子液体培养后提质粒进行酶切,酶切后跑核酸电泳验证,如图2所示。
65.实施例2构建敲除moae,gshb,ycea,ychj基因的重组大肠杆菌bl21δmoae,bl21δgshb,bl21δycea和bl21δychj
66.参考文献(jiang,y,et al.,multigene editing in the escherichia coli genome via the crispr-cas9 system.)公开的方法,运用crispr-cas9技术将大肠杆菌基因组上的moae基因、gshb基因、ycea基因和ychj基因敲除,改造后的菌株分别命名为bl21δmoae、bl21δgshb、bl21δycea和bl21δychj。
67.具体步骤包括:
68.2.1修复模板的获得
69.以大肠杆菌bl21基因组作为模板,取目的基因上下游各500bp设计引物进行pcr,产物胶回收后通过overlap pcr连接起来,引物核苷酸序列见seq id no.12-27,pcr条件同实施例1中步骤1.2。
70.2.2构建ptarget-moae质粒、ptarget-gshb质粒、ptarget-ycea质粒、ptarget-ychj质粒
71.a.以ptargetf质粒为模板,采用引物对moae-n20、gshb-n20、ycea-n20、ychj-n20进行反向pcr,引物核苷酸序列见seq id no.28-35,得到长度约2.1kb的dna片段。
72.moae-n20:catccccggatagtgttcga
73.gshb-n20:cgttaagtcagagaaccagg
74.ycea-n20:tcgcccaaaaacattcagcg
75.ychj-n20:gcatccctcttgtggagcag
76.pcr体系(100μl分装为5管):2
×
ps buffer 50μl,dntp 20μl,上、下游引物各3μl,模板1μl,kod酶1μl,dd h2o 21μl
77.pcr条件:95℃,10min;95℃,10s;55℃,15s;72℃,2min 30s(延伸时间根据片段长度而变化),38个循环。
78.b.用dpn i酶对pcr产物按照体系:pcr产物7μl,dpn i酶1μl,10
×
bufferμl,ddh2o 1μl进行原始模板的消化后进行原始模板的消化后转入trans1 t1感受态,涂布链霉抗性lb平板,37℃培养过夜,次日挑菌落接试管保菌即可。
79.2.3重组大肠杆菌bl2δmoae,、bl21δgshb、bl21δycea和bl21δychj的获得
80.a.通过热激将质粒pcas转化至宿主菌bl21(de3),涂在含有50μg/ml卡那霉素抗性的平板,30℃进行筛选。
81.b.挑取阳性克隆子,参考文献(jiang,y,et al.,multigene editing in the escherichia coli genome via the crispr-cas9 system.)制备电转感受态细胞bl21-cas9,制作过程加入10mm的阿拉伯糖诱导red的表达。
82.c.将步骤2.2中得到的质粒和步骤2.1中所得的修复模板片段电转至bl21-cas9感受态细胞中,30℃复苏2小时后,用硫酸卡那霉素(50μg/ml)+硫酸链霉素(40μg/ml)的平板进行筛选。
83.d.30℃培养20h后通过菌落pcr对克隆子进行验证,菌落pcr验证结果如图3所示,验证引物的核苷酸序列为seq id no.66-73。
84.e.阳性克隆子接lb液体(卡那抗性),加入0.5mm的iptg于30℃培养8-20小时,在卡那抗性的平板上划线分单菌落,并验证质粒ptarget的消除。质粒ptarget消除的阳性菌可重新进行下轮的基因编辑。
85.f.37℃液体培养,在无抗的平板上划线分单菌落,点板或液体培养验证质粒pcas的消除,得到基因敲除后的重组大肠杆菌(无抗),以该大肠杆菌为宿主,进行人表皮生长因子的表达生产。
86.实施例3构建整合表达bcsb,csgacsgb,fimh基因的重组菌(以整合至moae敲除位点为例)bl21-bcsb*,bl21-csgacsgb*,bl21-fimh*
87.3.1构建修复模板整合片段
88.a.以大肠杆菌基因组为模板进行目的基因片段pcr,以质粒pbbe1a为模板进行trc启动子pcr,以大肠杆菌mg1655基因组为模板进行目的基因片段pcr,回收pcr产物通过overlap pcr连接构成表达盒,引物核苷酸序列见seq id no.36-65。
89.b.以大肠杆菌基因组为模板,选取敲除位点基因上下游同源臂,进行pcr后将产物进行胶回收。
90.c.以胶回收的产物(上下同源臂和表达盒)作为模板overlap pcr进行连接。
91.3.2构建突变sgrna质粒:即敲除moae使用的ptarget质粒。
92.3.3整合片段的转化与转化子的筛选:同实施例2,菌落pcr验证结果如图4所示,验证引物的核苷酸序列为seq id no.66-67。
93.表1引物列表
94.95.[0096][0097]
实施例4 96孔板结晶紫染色法表征生物膜形成效果
[0098]
4.1具体实验步骤
[0099]
a.将实施例1~3所构建的重组菌株以及相应的对照菌大肠杆菌bl21(de3)在lb培养基中(通过质粒表达目的基因的菌株需添加100μg/ml的氨苄抗性)37℃、200rpm培养至对数期,再用无菌水稀释菌液至od
600
=0.1。
[0100]
b.96孔板中加入200μl的lb液体培养基(过表达菌株的培养基需添加iptg进行诱导),再将20μl稀释后菌液接入到培养基中,37℃下静置培养使大肠杆菌在96孔板底部成膜。
[0101]
c.之后倒掉lb液体培养基,用pbs冲洗2-3遍后,用甲醇4℃下固定15min后,倒掉晾干,加入1%的结晶紫染色15min;倒掉结晶紫染液,用pbs冲洗,加入200μl33%的冰醋酸并轻微震荡30min,使结晶紫脱色溶解,然后利用酶标仪在od
570
下读数,比较底部成膜生物量。
[0102]
d.把lb培养基换成lbg培养基(加入6g/l葡萄糖),重复上述的实验过程,观察菌株在加入葡萄糖的培养基中的成膜差异。
[0103]
4.2实验结果:如图5所示,通过质粒表达bcsb、csgacsgb、fimh三个基因可以明显促进96孔板底部成膜,整合表达这三个基因成膜效果不如质粒表达,但也在一定程度上促进了生物膜的形成。敲除gshb,ycea,ychj基因在lb培养基中没有明显的促进效果,但加入葡萄糖后发现成膜效果要优于对照。敲除moae基因后对生物膜的形成没有明显的促进作用。
[0104]
实施例5刚果红结合法表征生物膜形成效果
[0105]
5.1实验步骤
[0106]
a.挑实施例1~3所构建的重组菌株以及对照菌大肠杆菌bl21(de3)的单点至5ml lb液体(通过质粒表达目的基因的菌株需添加100μg/ml的氨苄抗性)培养过夜,取1%转接至5ml lb/lbg培养基(lbg培养基相较于lb培养基多加入6g/l葡萄糖)培养至对数生长期,加入0.5mm iptg诱导25℃培养20h(基因敲除菌不需要添加iptg诱导)。
[0107]
b.稀释到相同od值后,取2ml菌液离心,用pbs轻轻悬浮2次,离心。
[0108]
c.加入1ml pbs悬浮将菌体悬浮,加入刚果红(cr)染色(1g溶于100ml,加10μl),150rpm摇床,25℃反应10min,离心后观察沉淀及上清的颜色并对上清进行全波长扫描读取od=485nm处数值。
[0109]
5.2实验结果:质粒表达bcsb、csgacsgb、fimh三个基因的重组菌与刚果红发生了
明显的结合,在培养基中加入葡萄糖后结合效果更加明显。整合表达bcsb、csgacsgb、fimh,敲除moae、gshb、ycea、ychj后未观察到明显的与刚果红结合的现象,加入葡萄糖后,结合效果要略优于对照。
[0110]
刚果红结合率=1-od
485
/od
485(pbs+cr)
,改造菌株的刚果红结合率如表2所示。
[0111]
表2
[0112][0113]
实施例6 sds-page凝胶电泳检测蛋白条带及相对定量
[0114]
6.1蛋白电泳样品制备:取1ml发酵液至1.5ml离心管,测定od600后8000rpm离心2min,取上清21μl加入7μl 4*sds loading buffer充分混匀后于95℃水浴5min。
[0115]
6.2制胶:按照生工tricine-sds-page gel preparation kit试剂盒进行16.5%蛋白胶的配制。
[0116]
6.3上样:将蛋白胶板安装至垂直电泳槽中,加入电泳缓冲液。吸取20μl的样品加入样品孔中,并在相应孔位加入蛋白maker。
[0117]
电泳缓冲液配制:4
×
buffer 35ml+纯水105ml+10%sds 1.4ml
[0118]4×
buffer配制:tris 6g+甘氨酸28.8g+纯水500ml
[0119]
6.4电泳:接通电源,调节电压至120v,根据蛋白样品大小调整电泳时间。
[0120]
6.5染色及脱色:将电泳完毕的蛋白胶取出染色1h然后取出加入脱色液脱色直至背景颜色全部消失,倒去脱色液,使用凝胶成像仪对蛋白胶进行拍照。
[0121]
6.6以50mg/l hegf标品或250mg/l bsa为对照,使用image lab对蛋白胶目的条带进行相对定量分析后用菌体光密度od
600
数值进行折算。
[0122]
实施例7单批次游离发酵
[0123]
7.1活化:将甘油菌(实施例1~3所构建的重组菌株以及对照菌大肠杆菌bl21(de3))接种于5ml lb培养基(加入相应抗性,单质粒菌株加入50μg/l硫酸卡那霉素抗性,双质粒菌株加入50μg/l硫酸卡那霉素抗性和100μg/l氨苄抗性),接种量10μl,于37℃摇床中培养12h,转速200rpm。
[0124]
7.2接种:将培养基分装于500ml摇瓶灭菌,装液量100ml,然后加入相应抗性,接种量为1%,摇床转速200rpm,37℃培养。
[0125]
7.3诱导:待od
600
=0.6-0.8,加入1mm的iptg于25℃诱导48h,即得含有产物hegf的
发酵液。
[0126]
7.4产物检测:检测方式见实施例6,sds-page电泳结果显示如图6所示,通过软件定量分析后根据od
600
折算出相对hegf产量提升的倍数,如表3所示。
[0127]
表3
[0128][0129]
实施例8不同iptg浓度诱导下单批次游离发酵
[0130]
8.1方法同实施例7,所不同是发酵中研究三种不同iptg浓度:0.2mm,0.5mm,1mm对产人表皮生长因子效率的影响
[0131]
8.2实验结果:通过软件定量分析后根据od
600
折算出相对hegf产量提升的倍数,如表4所示。
[0132]
表4
[0133][0134]
实施例9不同培养基中单批次游离发酵
[0135]
9.1方法同实施例7,所不同的是发酵中研究三种不同培养基成分对产人表皮生长因子效率的影响,
[0136]
a.lb培养基:胰蛋白胨10g/l,酵母提取物5g/l,氯化钠10g/l;
[0137]
b.lbg培养基:胰蛋白胨10g/l,酵母提取物5g/l,氯化钠10g/l,葡萄糖5g/l;
[0138]
c.tb培养基:胰蛋白胨12g/l,酵母提取物24g/l,磷酸氢二钠9.4g/l磷酸二氢钾2.2g/l,甘油5ml/l。
[0139]
9.2实验结果:通过软件定量分析后根据od
600
折算出不同培养基中相对于对照菌
株hegf产量提升的倍数,如表5所示。
[0140]
表5
[0141][0142]
实施例10固定化连续发酵
[0143]
10.1活化、接种、诱导方式同实施例7,所不同的是接种时要在摇瓶中加入固定化载体(棉纤维,40g/l)进行固定化连续发酵。
[0144]
10.2在每一批发酵结束时全部放出发酵液,并同时补充新鲜发酵培养基,持续8个批次。
[0145]
10.3每隔12h取样测定发酵液中od
600
数值,结果如图9所示。从图中可以发现通过质粒表达bcsb基因和csgacsgb基因的细胞密度曲线与对照相比有些不同,在第四和第五批次中观察到发酵液中的细胞密度明显下降。而整合表达bcsb基因在第二和第三批次中发酵液的细胞密度也明显降低。这可能表明bcsb和csgacsb基因的表达有效地促进了细胞外基质的合成,使细胞能更好地吸附在载体上。
[0146]
10.4产物检测:取每一批发酵最末时间的发酵液跑蛋白电泳进行产物浓度相对定量分析,方法同实施例6,所不同的是不需要通过od
600
数值进行折算。发酵液中hegf sds-page凝胶电泳结果如图7所示,通过软件定量分析后相对hegf相对定量分析的结果如图8所示,产量提升的倍数如表6所示。
[0147]
表6
[0148]
[0149]
实施例11使用不同材料作为固定化载体进行固定化连续发酵
[0150]
方法同实施例10,所不同是固定化连续发酵中研究五种不同载体的使用对固定化连续发酵产人表皮生长因子效率的影响。
[0151]
五种固定化载体分别为棉纤维,无纺布,活性炭,聚酯纤维,尼龙纤维.
[0152]
通过软件定量分析后相对hegf产量提升的倍数如表7所示。
[0153]
表7
[0154][0155]
实施例12使用不同用量的棉纤维进行固定化连续发酵
[0156]
方法同实施例10,所不同是固定化连续发酵中研究五种不同棉纤维使用量对固定化连续发酵产人表皮生长因子效率的影响,分别是10g/l,20g/l,30g/l,40g/l,50g/l。
[0157]
通过软件定量分析后相对hegf产量提升的倍数如表8所示。
[0158]
表8
[0159][0160]
本发明提供了一种提高人表皮生长因子分泌生产效率的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1