一种比率光声型探针及其制备方法和在检测射线辐射剂量中的应用
1.本发明属于检测技术和成像领域,特别涉及一种比率光声型探针及其制备方法和在检测射线辐射量中的应用。
背景技术:
2.随着抗肿瘤药物种类的增多,对肿瘤治疗疗效的准确评价成为提高肿瘤治愈率的重要途径。因此,快速、无创、实时地检测和示踪肿瘤的凋亡过程,有助于对肿瘤的治疗疗效和药物的抗肿瘤活性作出准确的评价。荧光检测和成像技术近年来被广泛地应用于肿瘤检测与成像,然而,由于生物组织对光存在强的吸收和散射,活体荧光成像受到组织穿透深度低和空间分辨率低等限制,很难满足尤其是深部肿瘤的成像研究。为了克服光学成像的弊端,正电子发射断层成像(pet)技术由于具有较高的灵敏度和强的组织穿透力,被报道用于深部肿瘤的活性检测及成像研究。然而,由于pet示踪剂存在辐射性和有限空间分辨率的问题,阻碍了其在肿瘤凋亡可视化研究方面的广泛应用。mri造影剂虽然具有较高的分辨率和无限的穿透深度,但其敏感性较差,很难对肿瘤治疗早期进行准确性检测。众多抗肿瘤方案中,放疗占据重要的地位,射线剂量是治疗方案的重要参考,现有技术针对肿瘤诊断与治疗的探针无法对射线在肿瘤或者组织部位的剂量进行检测。因此,需要研发无创、高灵敏的新型分子探针,为肿瘤的治疗和预后评估治提供更为可靠的生物信息,为制定合适的诊疗方案提供重要的理论依据。
技术实现要素:
3.为克服现有技术的缺点与不足,本发明提供了一种比率光声型探针及其制备方法和应用,除了可以对放化疗过程中肿瘤细胞的凋亡过程和程度进行近红外和光声双模态成像,更主要的是,对于射线在肿瘤或者正常组织处的辐射剂量,利用本发明探针可进行实时无创的定性与定量分析。
4.为了实现上述目的,本发明的技术方案为:一种比率光声型探针,其结构式如下:上述比率光声型探针的制备方法,包括以下步骤:
(1)将化合物1与5-氯戊炔反应,得到化合物2;(2)将化合物2与原料a反应,得到化合物3;(3)将化合物3与间硝基苯酚反应,然后进行胺基化反应,得到化合物4;(4)将化合物4与原料b反应,得到化合物5;(5)将化合物5与rgd-n3反应,得到化合物6,即比率光声型探针。
5.进一步的,上述比率光声型探针的制备方法,包括以下步骤:(1)将化合物1与5-氯戊炔在金属卤素化合物存在下、有机溶剂中90~120℃反应10~15小时,得到化合物2;金属卤素化合物选自卤化钾,有机溶剂选自乙腈等;(2)将化合物2与原料a在混合溶剂中90~130℃反应5~12小时,得到化合物3;混合溶剂为醇、苯混合溶剂;(3)将化合物3与间硝基苯酚在无机碱、有机溶剂存在下室温反应8~15小时,然后在锡盐、酸存在下60~100℃反应10~15小时进行胺基化反应,得到化合物4;无机碱为无机钾盐,有机溶剂选自乙腈等;锡盐为无机系混合物,酸为浓盐酸;(4)将化合物4与原料b在hatu、dipea、有机溶剂存在下反应,反应结束后纯化,得到固体反应物,再将固体反应物溶入二氯甲烷中,加入tfa,得到化合物5;有机溶剂为二氯甲烷;(5)将化合物5与rgd-n3在有机溶剂、无机铜化合物、还原剂存在下室温反应5~10小时,得到化合物6,即比率光声型探针;有机溶剂为砜溶剂,无机铜化合物为硫酸铜,还原剂为抗坏血酸钠。
6.作为示例,上述比率光声型探针的制备方法,包括以下步骤:(1)分别将化合物1和5-氯戊炔溶于乙腈中,向其中加入ki,封管100℃加热12 h,纯化后得到化合物2;(2)将化合物2和原料a溶入正丁醇与甲苯中,110℃加热8h,纯化后得到化合物3;(3)将间硝基苯酚和碳酸钾溶于乙腈中,室温下搅拌15分钟,然后向其中加入化合物3,室温继续搅拌12 h,纯化得到固体反应产物;然后将上述固体反应产物与氯化亚锡、甲醇、浓盐酸混合,然后80℃回流12h,然后纯化得到化合物4;(4)将原料b溶于二氯甲烷中,向其中加入hatu和dipea,室温搅拌30 min,然后向其中加入化合物4,室温搅拌48 h,hplc纯化;将纯化后的产物溶入二氯甲烷中,加入tfa,室温反应15 min,然后纯化得到化合物5;(5)将化合物5和rgd-n3溶于dmso中,向其中加入五水硫酸铜和抗坏血酸钠混合后的溶液,室温搅拌8 h,然后纯化后得到化合物6,即为该比率光声型探针acdevd-cy-rgd。
7.上述技术方案中,步骤(1)所述的原料1、5-氯戊炔、ki的当量比为1∶1.5∶(1.5~2.5),优选为1:1.5:2;步骤(2)所述的化合物2和化合物a的当量比为2:1,所述的溶剂正丁醇与甲苯两者的体积比为7:1;步骤(3)所述的化合物3、间硝基苯酚、碳酸钾、氯化亚锡的当量比为1∶2.5∶(2~3)∶(4~8),优选为1:2.5:2.5: (5.5~6),所述的浓盐酸与甲醇的体积比为1:8;步骤(4)所述的化合物4、原料b、hatu和dipea的当量比为1∶2.5∶(1~2)∶(1.5~2.5),优选为1:1.1:1.5:2,所述的三氟乙酸(tfa)与二氯甲烷的体积比为1:2;步骤(5)所述的化合物5、rgd-n3、cuso4·
5h2o、抗坏血酸钠的当量比为1∶1∶(1.5~2.5)∶(2~2.5),优选为1:1:2:2.2。
8.本发明中,rgd-n3的化学结构式如下:本发明中,化合物1、化合物2、化合物3、化合物4、化合物5、原料a、原料b的化学结构式分别如下:本发明中,原料a、原料b的化学结构式分别如下:本发明化合物2、化合物3、化合物4、化合物5、化合物6(acdevd-cy-rgd,比率光声
型探针)都是离子形式,为本领域常规表示方法,常用卤素阴离子配位,例如i-,cl-等,具体配位方法也为常规技术,不影响本发明技术效果的实现。
9.本发明公开了上述比率光声成像探针在制备荧光成像试剂或者光声成像试剂或者比率光声成像试剂中的应用;或者上述比率光声成像探针在检测射线辐射剂量中的应用;或者上述比率光声成像探针在制备检测射线辐射剂量试剂中的应用。具体的,射线辐射可以在体外,也可在体内,可以在肿瘤部位,也可在正常组织部位。
10.本发明公开了一种检测射线辐射剂量的方法,射线辐射后,将上述比率光声成像探针置于射线辐射部位,然后检测与计算探针在680和710 nm激发光下光声信号的强度增量,分别记为δpas
680
、δpas
710
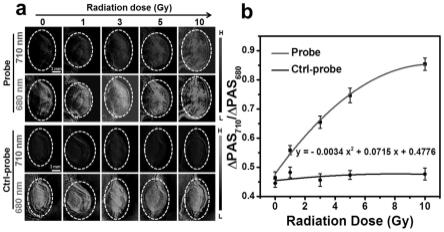
,将δpas
710
/δpas
680
的数值带入δpas
710
/δpas
680
比率-射线辐射剂量关系曲线,得到射线辐射剂量。优选的,射线辐射后,48小时后,将上述比率光声成像探针置于射线辐射部位,8小时后检测与计算探针在680 和710 nm激发光下光声信号的强度增量。本发明首次给出射线辐射剂量的定量检测方法,解决了现有技术只能定性观察射线辐射结果的问题。将比率光声成像探针置于射线辐射部位的具体方法为常规方法,可以为注射,也可以为吞服或者雾化,还可以为涂抹,与辐射部位有关。
11.本发明中,用于定量检测射线辐射剂量的δpas
710
/δpas
680
比率-射线辐射剂量关系曲线为,其中y为δpas
710
/δpas
680
比率,x为射线辐射剂量。
12.由于上述技术方案的运用,本发明的优点是:1. 本发明设计合成了一种比率光声探针,其最大激发和发射位于近红外区域,穿透力强,背景干扰小,可用于活体组织或者体外射线辐射以及辐射剂量的检测。
13.2. 本发明中的比率光声探针可对放化疗过程中肿瘤细胞的凋亡过程和程度进行近红外荧光和光声双模态成像。
14.3. 本发明中比率光声探针可对射线辐射剂量进行实时无创的成像与定量分析,首次实现对活体肿瘤或者组织部位的射线辐射剂量检测。
附图说明
15.图1为比率光声探针的合成示意图。
16.图2为对照光声探针的合成示意图。
17.图3为实施例2中(a)比率光声探针acdevd-cy-rgd尾静脉注射入未治疗(0 gy)或放疗(1、3、5或10 gy)4t1荷瘤小鼠不同时间点近红外荧光成像。(b)对应(a)中荧光强度随时间变化。
18.图4为实施例3中(a)比率光声探针acdevd-cy-rgd尾静脉注射入未治疗(0 gy)或放疗(1、3、5或10 gy)4t1荷瘤小鼠不同时间点的光声成像,(b)对应(a)中比率光声信号(δpas
680
)随时间变化,(c)对应(a)中比率光声信号(δpas
710
)随时间变化,(d)对应(a)中8小时时间点的比率光声信号(δpas
710
/δpas
680
)随辐射剂量变化;*p 《 0.05 (n = 3), **p 《 0.01 (n = 3), ***p 《 0.001 (n = 3)。
19.图5为实施例4中(a)比率光声探针acdevd-cy-rgd或者对照光声探针accy-rgd尾静脉注射入未治疗(0 gy)或放疗(1、3、5或10 gy)4t1荷瘤小鼠8 h时间点的光声成像;(b)8小时时间点的比率光声信号(δpas
710
/δpas
680
)与辐射剂量的关系曲线。
20.图6为使用关系曲线定量辐射剂量。(a) 由未知辐射剂量诱导的4t1肿瘤凋亡的pa成像示意图(小鼠被1.5或2.5 mm厚的铝板覆盖,然后向肿瘤局部输送规定剂量的10 gy x射线)。(b)注射acdevd cy rgd(1.5 μg g-1
)后8小时,在680和710 nm(比例尺=2 mm)处收集接受未知剂量x射线照射的4t1肿瘤的pa图像。(c) 肿瘤区域在680和710 nm的pa强度增量(δpas)。(d)不同铝板厚度下δpas
710
/δpas
680
的比值。(e)不同铝厚度下的测量辐射剂量。(f)测量剂量和计算剂量的结果。
具体实施方式
21.下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。荷瘤的balb/c雌性小鼠为常规方法建模,且符合苏州大学动物实验要求。
22.实施例1 本发明所述的比率光声型探针的制备方法,合成步骤如图1所示,化合物中,配位阴离子为碘离子,具体如下:(1)将1.54 g的5-氯戊炔(15 mmol)溶于50 ml乙腈中,向其中加入4.98 g的ki(30 mmol),加热回流30 min,然后向其中加入2.09 g的化合物1(10 mmol),加热回流48 h,反应结束后降至室温,旋干,加水,二氯甲烷萃取3次,无水硫酸钠干燥,旋至二氯甲烷剩余5 ml,向其中加入100 ml乙醚,有大量灰绿色的固体析出,过滤,乙醚洗涤3次,抽干,得到1.29 g灰绿色的固体粉末,即为化合物2,产率为32%。1h nmr (600 mhz, dmso-d6) δ 8.38 (d, j = 8.4 hz, 1h), 8.31 (d, j = 8.9 hz, 1h), 8.22 (d, j = 8.2 hz, 1h), 8.15 (dd, j = 8.9, 1.8 hz, 1h), 7.79 (t, j = 7.5 hz, 1h), 7.73 (t, j = 7.5 hz, 1h), 4.62 (t, j = 6.4 hz, 2h), 3.01
–
2.92 (m, 4h), 2.46 (t, j = 7.0 hz, 2h), 2.18
–
2.09 (m, 2h), 1.77 (s, 6h). 13
c nmr (151 mhz, dmso-d6) δ 197.81, 139.47, 137.88, 133.96, 131.63, 130.66, 129.37, 128.22, 128.17, 124.37, 114.04, 84.06, 73.44, 56.54, 48.02, 27.20, 22.52, 16.18, 14.87. calcd. for c
20h22n+
, ([m]
+
): 276.1747, found esi-ms: m/z 276.1741。
[0023]
(2)将806 mg的化合物2(2 mmol)和172 mg的2-氯-3-(羟亚甲基)-1-环己烯-1-羧醛(1 mmol,原料a)溶于正丁醇(7 ml)与甲苯(1ml)中,加热至110℃,回流8 h,反应结束后将溶剂旋干,用甲醇与乙醚重结晶,过滤,乙醚洗涤2次,得578 mg深褐色固体,即为化合物3,产率为71%。1h nmr (600 mhz, dmso-d6) δ 8.39 (d, j = 14.1 hz, 2h), 8.31 (d, j = 8.5 hz, 2h), 8.12 (d, j = 8.8 hz, 2h), 8.08 (d, j = 8.2 hz, 2h), 7.79 (d, j = 8.9 hz, 2h), 7.69
–
7.64 (m, 2h), 7.56
–
7.51 (m, 2h), 6.41 (d, j = 14.2 hz, 2h), 4.39 (t, j = 7.2 hz, 4h), 3.04
–
2.97 (m, 2h), 2.80
–
2.72 (m, 4h), 2.42
–
2.35 (m, 4h), 1.97 (s, 12h). 13
c nmr (151 mhz, dmso-d6) δ 174.50, 148.47, 143.09, 140.61, 134.57, 132.44, 131.42, 130.88, 128.77, 128.40, 127.30, 126.02, 123.25, 112.51, 102.08, 84.50, 73.27, 51.73, 43.97, 40.99, 27.99, 27.17, 26.95, 16.25. calcd. for c
48h48
cln
2+
, ([m]
+
): 687.3501, found esi-ms: m/z 687.3489。
[0024]
(3)将458 mg间硝基苯酚(2.5 mmol)溶于10 ml乙腈中,加入345 mg碳酸钾(2.5 mmol),室温的条件下搅拌15 min,然后将814 mg化合物5(1.0 mmol)加入其中,室温的条件
下继续搅拌8 h,待反应结束后,加水、二氯甲烷萃取3次,无水硫酸钠干燥,旋干,得到深褐色固体,直接用于投下一步。
[0025]
(4)将上一步旋干物和1128 mg氯化亚锡溶于10 ml甲醇中,向其中加入2 ml的浓hcl(37wt%),然后80℃回流12 h,待反应结束后,加入饱和的碳酸钠溶液调节ph = 7.5,过滤掉固体残渣,二氯甲烷萃取滤液3次,无水硫酸钠干燥,旋干,过硅胶柱纯化(二氯甲烷:甲醇=10:1),得到330 mg深蓝色固体,即为化合物4(cynh2),上述两步产率为56%。1h nmr (400 mhz, dmso-d6) δ 8.57 (d, j = 14.4 hz, 1h), 8.24 (d, j = 8.5 hz, 1h), 8.13 (d, j = 8.9 hz, 1h), 8.09 (d, j = 8.1 hz, 1h), 7.80 (d, j = 8.9 hz, 1h), 7.74
–
7.64 (m, 2h), 7.55 (t, j = 7.3 hz, 1h), 7.43 (d, j = 8.5 hz, 1h), 6.79 (s, 2h), 6.78
–
6.71 (m, 2h), 6.34 (d, j = 14.4 hz, 1h), 4.39 (t, j = 7.2 hz, 2h), 2.99 (t, j = 2.5 hz, 1h), 2.77
–
2.66 (m, 4h), 2.39 (td, j = 6.8, 2.5 hz, 2h), 2.03
–
1.94 (m, 8h), 1.90
–
1.79 (m, 2h). 13
c nmr (151 mhz, dmso-d6) δ 175.77, 163.27, 156.64, 155.93, 141.92, 140.50, 139.36, 134.30, 132.45, 131.40, 130.94, 130.78, 128.79, 128.29, 125.97, 123.46, 122.92, 115.53, 115.01, 113.85, 112.52, 100.48, 98.30, 84.43, 73.23, 51.74, 49.51, 43.91, 28.85, 28.48, 27.06, 21.16, 16.21. calcd. for c
34h33
n2o
+
, ([m]
+
): 485.2587, found esi-ms: m/z 485.2573。
[0026]
(5)将15.1 mg的原料b(0.022 mmol)溶于6 ml二氯甲烷中,加入6.6 μl的dipea(0.04 mmol,n,n-二异丙基乙胺)和11.4 mg的hatu(0.03 mmol,2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐),室温搅拌15 min,然后向其中加入12.3 mg的化合物4(0.05 mmol),室温条件下继续搅拌24 h,待反应结束后,旋去二氯甲烷,得蓝色固体,直接用于投下一步。
[0027]
(6)将上述蓝色固体溶于3 ml二氯甲烷中,向其中缓慢的滴加1 ml三氟乙酸,室温搅拌30 min,待反应结束后,将反应旋干,hplc制备柱制备得到12.7 mg化合物5,上述两步产率为57%。1h nmr (400 mhz, dmso-d6) δ 10.44 (d, j = 47.3 hz, 1h), 8.77
–
8.55 (m, 1h), 8.53
–
8.31 (m, 1h), 8.27
–
8.20 (m, 2h), 8.19
–
8.08 (m, 1h), 8.03
–
7.94 (m, 2h), 7.88 (dd, j = 26.5, 8.0 hz, 1h), 7.76 (t, j = 7.6 hz, 1h), 7.69
–
7.62 (m, 1h), 7.61
–
7.54 (m, 1h), 7.53
–
7.43 (m, 2h), 6.66 (d, j = 15.1 hz, 1h), 4.85
–
4.71 (m, 1h), 4.63
–
4.55 (m, 2h), 4.37
–
4.28 (m, 1h), 4.21
–
4.09 (m, 1h), 3.00 (t, j = 2.5 hz, 1h), 2.89
–
2.79 (m, 1h), 2.79
–
2.68 (m, 3h), 2.67
–
2.57 (m, 2h), 2.47
–
2.41 (m, 2h), 2.23
–
2.14 (m, 2h), 2.1
–
1.94 (m, 5h), 1.85 (t, j = 6.5 hz, 5h), 1.43
–
1.33 (m, 12h), 1.33
–
1.31 (m, 3h), 1.23 (s, 6h). calcd. for c
54h61
n6o
12+
, ([m]
+
): 985.4342, found esi-ms: m/z 985.4346。
[0028]
(7)将11.1 mg化合物5(0.01 mmol)和6.9 mg的rgd-n3(0.011mmol)溶于1ml的dmso中,然后将400 μl 五水硫酸铜溶液(0.05 m)与440 μl抗坏血酸钠溶液(0.05 m)混合,超声五分钟后滴入到上述溶液中,室温条件下搅拌5 h,待反应结束后,hplc制备柱制备得到13.4 mg化合物6,即为该比率光声型探针acdevd-cy-rgd,产率为76%。calcd. for c
81h100n17o20+
, ([m]
+
): 1630.7325, found esi-ms: m/z 1630.7314。
[0029]
对照探针的制备如图2。将cynh2(61.2 mg,0.1 mmol)溶于6 ml二氯甲烷中,室温
的条件下向其中加入乙酸酐(20 μl,0.2 mmol),室温继续搅拌5 h。旋去反应溶剂,使用制备型高效液相色谱仪分离纯化,得到56.2 mg的化合物7,产率为86%。1h nmr (600 mhz, cd3od) δ 8.90 (d, j = 15.0 hz, 1h), 8.38 (d, j = 8.5 hz, 1h), 8.18 (s, 1h), 8.14 (d, j = 8.8 hz, 1h), 8.09 (d, j = 8.2 hz, 1h), 7.82 (d, j = 8.9 hz, 1h), 7.75 (t, j = 7.6 hz, 1h), 7.62 (t, j = 7.5 hz, 1h), 7.46 (d, j = 8.4 hz, 1h), 7.34 (s, 1h), 7.24 (dd, j = 8.4, 1.8 hz, 1h), 6.66 (d, j = 15.0 hz, 1h), 4.60 (t, j = 7.5 hz, 2h), 2.83-2.78 (m, 2h), 2.76 (t, j = 6.0 hz, 2h), 2.58 (t, j = 2.5 hz, 1h), 2.47 (td, j = 6.5, 2.5 hz, 2h), 2.22 (s, 3h), 2.20-2.13 (m, 2h), 2.12 (s, 6h), 2.00-1.92 (m, 2h). 13
c nmr (151 mhz, cd3od) δ 181.05, 172.26, 162.70, 155.10, 146.74, 143.79, 140.37, 137.54, 134.57, 134.23, 132.50, 131.44, 130.11, 129.43, 129.38, 129.18, 127.60, 123.88, 119.44, 118.00, 115.99, 112.86, 107.20, 104.96, 83.83, 72.00, 54.03, 45.48, 30.29, 28.28, 27.97, 25.34, 24.35, 21.73, 16.68. calcd. for c
36h36
n2o
22+
, ([m+h]
+
): 528.2766, found esi-ms: m/z 528.2740。将化合物7(13.8 mg, 0.02 mmol)和rgd-n3(13.8 mg, 0.022 mmol)溶于1ml的二甲基亚砜中。然后,将五水硫酸铜溶液(800 μl,0.05 m)与抗坏血酸钠溶液(880 μl,0.05 m)混合,超声五分钟后滴入到上述反应液中,室温条件下搅拌6 h,待反应结束后,通过制备型高效液相色谱仪(hplc)分离纯化,得到22.7 mg化合物8,即为对照组探针accy-rgd,产率为88%。calcd. for c
63h75n13o102+
, ([m+h]
+
): 1173.5749, found esi-ms: m/z 1173.5688。
[0030]
实施例2 比率光声探针的荧光成像分别以单次0,1,3,5,10 gy 剂量x射线照射荷瘤(4t1小鼠乳腺癌)的balb/c雌性小鼠的肿瘤。停止照射48小时后,将实施例1获得的比率光声探针acdevd-cy-rgd溶于pbs中(1.5 μg g-1
),以尾静脉的方式将其注入小鼠体内,随后置于小动物成像系统(ivis)中(激发波长:710 nm)进行实时荧光成像,然后通过ivis系统软件计算小鼠肿瘤部位不同时间点的荧光强度。结果如图3所示,荧光信号在注入探针后8小时达到最大值,然后随着时间的推移而减小。同时,随着x射线剂量的增大,荧光信号逐渐的增强,对于接受10 gy x射线照射的肿瘤,相对于未接受放疗的对照肿瘤,fl增强约11.52倍。综上所述,这些证据充分证明了该探针在活体肿瘤凋亡的敏感检测和实时监测方面具有很大的潜力。
[0031]
实施例3 比率光声探针的光声成像分别以单次0,1,3,5,10 gy 剂量x射线照射荷瘤(4t1小鼠乳腺癌)的balb/c雌性小鼠的肿瘤。停止照射48小时后,将实施例1获得的比率光声探针acdevd-cy-rgd溶于pbs中(1.5 μg g-1
),以尾静脉的方式将其注入小鼠体内,利用msot在680 ~ 800 nm激发光下采集不同时间点的光声图像。 成像重建后,利用msot成像系统软件包对肿瘤区域的探针信号进行roi分析。为了减少肿瘤组织固有光声信号(pas)的干扰,在静脉注射探针后,记录随着时间推移光声信号的强度增量(δpas-通过减去注射探针前获得)。结果如图4所示,x-ray辐照下,肿瘤部位在680 nm和710 nm处的光声强度呈现先增后减的变化趋势,并在8 h附近达到峰值。相较于680 nm处的光声,肿瘤部位在710 nm处的光声强度随着x-ray辐射剂量的增加而增加。同时,比率光声信号(δpas
710
/δpas
680
)也随着辐射剂量的增加而增加。
[0032]
实施例4 比率光声信号与x射线剂量的关系
分别以单次0,1,3,5,10 gy 剂量x射线照射荷瘤(4t1小鼠乳腺癌)的balb/c雌性小鼠的肿瘤。停止照射48小时后,将实施例1获得的比率光声探针acdevd-cy-rgd或者对照探针accy-rgd溶于pbs中(1.5 μg g-1
),以尾静脉的方式将其注入小鼠体内,利用msot在680 和710 nm激发光下采集8小时的光声图像。成像重建后,利用msot成像系统软件包对肿瘤区域的探针信号进行roi分析。为了减少肿瘤组织固有光声信号(pas)的干扰,在静脉注射探针后,记录随着时间推移光声信号的强度增量(δpas,通过减去注射探针前的光声信号获得),acdevd-cy-rgd的实验与实施例3一致,为其8小时的结果。结果如图5所示,放疗肿瘤中探针的比率光声信号(δpas
710
/δpas
680
)随x射线剂量增加而增强;静脉注射对照探针accy-rgd后,对不同剂量x射线照射的肿瘤均未检测到明显的比率光声信号(δpas
710
/δpas
680
)增强,见图5a。基于上述结果,对比率光声信号与辐射剂量进行拟合,并建立非线性关系公式,为精确评估辐射剂量提供卓有成效的工具,见图5b,对照探针的比率光声信号不随x射线剂量增加而增强。
[0033]
实施例5 比率光声探针用于辐射剂量的定量评估准确评估肿瘤及其周围组织的辐射水平,对于优化临床治疗方案,最大限度地减少辐射副反应具有重要意义。如图6a所示,将1.5或2.5 mm厚的铝板覆盖在雌性balb/c小鼠4t1皮下瘤上,随后给予10 gy剂量的x射线照射肿瘤部位,以此作为模拟未知辐射剂量的肿瘤模型。停止照射48 h后,将1.5 μg g-1
的探针acdevd-cy-rgd经尾静脉注射入小鼠体内,注射8 h后记录710 nm和680 nm通道的光声图像,肿瘤部位的δpas信号是通过减去注射探针前记录的肿瘤初始光声信号后获得。进一步计算比率光声信号(δpas
710
/δpas
680
)值,并分别代入到比率光声和辐射剂量的关系公式(实施例4)来获得两组的辐射剂量(计算剂量),为6.02
ꢀ±ꢀ
0.53 gy和4.42
ꢀ±ꢀ
0.40 gy,误差百分数分别约为7.67% 和8.49%(图6d,图6e)。同时,使用了一种常用的传统剂量计(rs2000pro (rad source, usa))来测量铝板下方的剂量值,作为测量剂量。通过比较计算剂量和测量剂量,惊奇地发现,本发明计算辐射剂量与实际剂量非常接近,而且非常准确。总之,这些结果强有力地证明了本发明比率光声成像探针在肿瘤放疗中具有精确评估辐射剂量的潜力。
[0034]
基于上述研究结果,本发明的探针acdevd-cy-rgd作为剂量计来评估活体肿瘤辐射剂量是可行的;进一步的,在组织收到射线照射后,根据δpas
710
/δpas
680
,可准确估算之前接收的射线照射量。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1