一种三足六脲中性配体、制备方法及其应用
1.本发明涉及一种三足六脲中性配体、制备方法及其应用,属于高分子材料技术领域。
背景技术:
2.阴离子在环境科学、生命科学、生物学和医学等研究领域扮演着重要的角色。例如,高浓度so
42-极易导致水体矿化、永久性硬度指标高,诱发工业水垢的形成;而no
3-和cl-等则是工业污染的主要成分。因此,阴离子识别和分离具有重要的研究和应用价值。现行废水处理方法存在成本高、选择性低、流程复杂的特点,难以应对实际生产需求。吴彪等报道了一种尾端为硝基的三足六脲中性配体,该配体可与硫酸根离子特异性结合,用于对硫酸根离子的识别。然而该配体中尾端的硝基基团易发生强化还原反应,在实际应用中具有一定的局限性;且受溶解度的影响,该配体的可重复利用率较低;同时该配体回收时采用bacl2沉淀法,引入重金属钡盐成本较高,且有较大污染环境风险。
技术实现要素:
3.有鉴于此,本发明的目的在于提供一种三足六脲中性配体、制备方法及其应用。
4.为实现上述目的,本发明的技术方案如下:
5.一种三足六脲中性配体,所述配体的结构式如下:
6.7.其中,r为-c
nh2n+1
、-phc
nh2n+1
或-phoc
nh2n+1
n为大于等于3的正整数。
8.优选的,n为6~18。
9.本发明所述的一种三足六脲中性配体的制备方法,所述方法步骤包括:
10.将对甲基苯异氰酸酯或叔丁基异氰酸酯溶于四氢呋喃(thf)中得到溶液ⅳ;氮气氛围下,将化合物ⅰ溶于四氢呋喃和甲苯的混合溶剂中得到溶液
ⅴ
并加热至110℃以上,逐滴滴加溶液ⅳ,通过薄层色谱分析检测化合物ⅰ反应完全,固液分离,固体洗涤、干燥,得到一种三足六脲中性配体;
11.或,将烷基胺溶于四氢呋喃中得到溶液ⅵ;氮气氛围下,将4-硝基苯氯甲酸酯溶于四氢呋喃中得到溶液ⅶ并加热至80℃以上,然后逐滴滴加溶液ⅵ,通过薄层色谱分析检测4-硝基苯氯甲酸酯反应完全,冷却后滤液浓缩,加入反溶剂ⅰ至沉淀析出完全,过滤,固体洗涤后干燥,得到中间产物a(氨基甲酸酯);将所述中间产物a溶于四氢呋喃中得到溶液
ⅷ
;氮气氛围下,将化合物ⅰ溶于四氢呋喃得到溶液
ⅸ
并加热至80℃以上,然后逐滴滴加溶液
ⅷ
,通过薄层色谱分析检测化合物ⅰ反应完全,冷却后,固液分离或浓缩加入反溶剂ⅱ至沉淀析出完全,过滤,固体洗涤后干燥,得到一种三足六脲中性配体;
12.其中,化合物ⅰ的结构式为:
13.烷基胺的化学式为nh
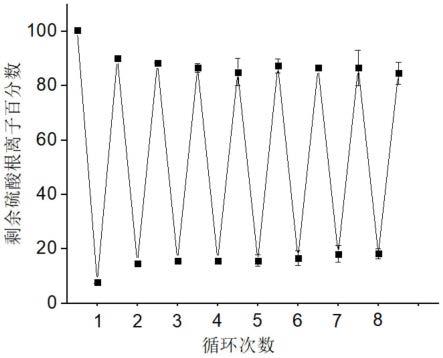
2-r,r为-c
nh2n+1
、-phc
nh2n+1
或-phoc
nh2n+1
,n为大于等于3的正整数;
14.中间产物a与化合物ⅰ的摩尔比大于等于3:1。
15.优选的,所述化合物ⅰ通过以下方法制备得到,所述方法步骤如下:(1)将邻硝基苯异氰酸酯溶于甲苯中得到溶液ⅰ,将三(2-氨基乙基)胺溶于四氢呋喃中得到溶液ⅱ,将溶液ⅰ逐滴滴加到溶液ⅱ中,通过薄层色谱分析(tlc)检测三(2-氨基乙基)胺反应完全,固液分离,固体洗涤、干燥,得到中间产物1;其中,邻硝基苯异氰酸酯与三(2-氨基乙基)胺的摩尔比≥3:1;
16.(2)将所述中间产物1溶于乙醇中得到溶液ⅲ,加入钯碳催化剂(pd/c)并加热至90℃以上,搅拌20分钟以上后,逐滴滴加还原剂水合肼到溶液ⅲ,通过薄层色谱分析检测中间产物1反应完全,收集滤液,旋蒸浓缩至有白色沉淀析出,静止沉淀析出完全,过滤后沉淀洗涤、干燥,得到化合物ⅰ。
17.优选的,所述溶液
ⅷ
中还加入有机碱催化剂三乙胺,三乙胺与中间产物a的摩尔比
≥1:1。
18.优选的,所述反溶剂ⅰ为正己烷或甲醇;所述反溶剂ⅱ为乙腈。
19.本发明所述的一种三足六脲中性配体的应用,所述三足六脲中性配体用于阴离子的识别。由于所述配体为中性配体,进行阴离子识别时在相转移催化剂如甲基三辛基氯化铵(a336cl)的作用下进行。
20.优选的,所述三足六脲中性配体作为萃取剂用于硫酸根离子的液液萃取。
21.优选的,所述三足六脲中性配体回收时,将所述三足六脲中性配体与硫酸根形成的萃合物,加入到ph小于1的盐酸或硝酸中,回收有机相,干燥后得到三足六脲中性配体。
22.优选的,所述三足六脲中性配体可重复使用八次以上。
23.有益效果
24.本发明提供了一种三足六脲中性配体,以三胺桥联基为骨架,可重复的邻苯二脲结构为基本单元,通过-c
nh2n+1
、-phc
nh2n+1
或-phoc
nh2n+1
尾端取代基组成三足六脲中性配体。该配体能够通过脲基官能团强氢键给体作用可与阴离子结合,形成配合物。阴离子与三足六脲萃取剂之间的氢键作用,末端取代基c-h
···
π相互作用,说明该三足六脲中性配体对阴离子具有识别能力。
25.本发明提供了一种三足六脲中性配体的制备方法,所述方法原料简单易得,具有广泛的普适性。其中一种方法是通过异氰酸酯与化合物ⅰ直接反应得到三足六脲中性配体;另一种方法是通过将胺类物质转化为氨基甲酸酯,再与化合物ⅰ反应得到三足六脲中性配体。
26.本发明提供了一种三足六脲中性配体的应用,该配体可用于对阴离子的识别,尤其是对硫酸根具有高萃取效率和高选择性(大于95%)。
27.本发明提供了一种三足六脲中性配体的释放回收方法,水相ph小于1时,游离于有机相和水相界面的硫酸根离子质子化,转变为硫酸氢根离子(hso
4-),其与配体的结合能力相比于硫酸根离子明显下降,从而被配体释放,进入水相。由此通过质子化实现与萃取剂结合的硫酸根离子的可控释放,并具有较高的可重复性;且8次循环后仍能保证80%以上的萃取效率。
附图说明
28.图1为对比例4中所述中性配体的萃取与反萃取结果图。
29.图2为对比例5中所述中性配体的萃取与反萃取结果图。
30.图3为实施例10中所述中性配体的萃取与反萃取结果图。
31.图4为实施例11中所述中性配体的萃取与反萃取结果图。
具体实施方式
32.下面结合具体实施例对本发明作进一步详细的说明。
33.实施例1
34.一种化合物ⅰ的制备方法,所述方法步骤如下:
35.(1)250ml两口瓶中将邻硝基苯异氰酸酯(5.0g,30.5mmol,3.2eq)溶于甲苯(80ml)中得到溶液ⅰ;25ml滴液漏斗中将三(2-氨基乙基)胺(1.4g,9.6mmol,1eq)溶于干燥thf
(10ml)中得到溶液ⅱ,以2秒/滴的速率将溶液ⅰ逐滴滴加到溶液ⅱ中,tlc检测三(2-氨基乙基)胺反应完全,生成产物点(比移值rf=0.6,展开剂:二氯甲烷(dcm)和甲醇(ch3oh)的体积比为50:2),反应结束,固液分离,固体依次用甲苯(10ml
×
3)和乙醚(10ml
×
3)洗涤后,真空干燥,得到中间产物1(5.9g,产率为98%);1h nmr(400mhz,298k,dmso-d6,ppm):δ=9.35(s,3h),8.24(d,3h,j=8.48hz),7.99(d,3h,j=9.84hz),7.57(t,3h,j=8.6hz),7.49(s,3h),7.08(t,3h,j=8.36hz),3.2(m,6h),2.63(t,6h,j=6.6hz)。
36.(2)1000ml两口瓶中将所述中间产物1(3.5g,5.5mmol,1eq)溶于乙醇(500ml)中,超声分散均匀,得到溶液ⅲ,加入钯碳催化剂(pd/c)(催化剂总质量为0.7g,pd的负载量为10wt%)并加热至90℃,搅拌20分钟后,以2秒/滴的速率逐滴滴加还原剂水合肼(8.5ml)到溶液ⅲ,tlc检测中间产物1反应完全,生成产物点(rf=0.6,展开剂:dcm和ch3oh的体积比为50:3),反应结束,砂芯漏斗上铺上滤纸,硅藻土过滤除去pd/c,滤液旋蒸浓缩至有白色沉淀析出,静止沉淀析出完全,过滤后沉淀用乙醚(10ml
×
3)洗涤后,真空干燥,得到化合物ⅰ(2.4g,产率为74%)。1h nmr(400mhz,298k,dmso-d6,ppm):δ=7.61(s,3h),7.21(d,3h,j=7.84hz),6.69(t,3h,j=7.8hz),6.51(d,3h,j=8.56hz),6.21(t,3h,j=8.56hz),4.69(s,6h),3.18(m,6h),2.58(t,6h,j=6.32hz)。
37.对比例1
38.一种三足六脲中性配体的制备方法,所述方法步骤如下:
39.125ml滴液漏斗中将对甲苯异氰酸酯(1.2ml,9mmol,3.5eq)溶解于干燥thf(20ml)得到溶液ⅳ;在氮气氛围下,500ml三口瓶中将实施例1所述化合物ⅰ(1.44g,2.6mmol,1.0eq)溶于干燥thf(20ml)和甲苯(180ml)的混合溶剂中得到溶液
ⅴ
并加热至110℃,然后以2秒/滴的速率逐滴滴加溶液ⅳ;反应14小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:二氯甲烷(dcm)和甲醇(ch3oh)的体积比为50:6),反应结束,冷却至室温后固液分离,固体分别用thf(10ml
×
3)和乙醚(10ml
×
3)洗涤、干燥,得到一种三足六脲中性配体(2g,产率为85%),记为hu-ph-ch3;1h nmr(400mhz,298k,dmso-d6,ppm):δ=8.96(s,1h),7.96(s,1h),7.91(s,1h),7.57(dd,j=7.6,2.0hz,1h),7.42(dd,j=7.6,2.1hz,1h),7.30(d,j=8.0hz,2h),7.06
–
6.93(m,4h),6.50(t,j=5.5hz,1h),3.19(d,j=6.4hz,2h),2.60(t,j=6.9hz,2h),2.22(s,3h)。
40.反应方程式如下:
[0041][0042]
实施例2
[0043]
一种三足六脲中性配体的制备方法,所述方法步骤如下:
[0044]
(1)125ml滴液漏斗中将己胺(1.91g,18.8mmol,1.0eq)溶解于干燥thf(60ml)得到溶液ⅵ;氮气氛围下,250ml三口瓶中将4-硝基苯氯甲酸酯(4.50g,22.4mmol,1.2eq)溶于干燥thf(60ml)中得到溶液ⅶ并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液ⅵ,反应14小时后,tlc检测4-硝基苯氯甲酸酯反应完全,生成产物点(比移值rf=0.7,展开剂:dcm),反应结束,体系澄清,减压浓缩至干,冷却至室温,加入正己烷(150ml),充分超声搅拌,至沉淀析出完全,过滤沉淀,正己烷(10ml
×
3)洗涤沉淀,干燥,得到中间产物a1(2.8g,产率为60%);1h nmr(400mhz,298k,chcl3,ppm):δ=8.25(dd,2h,j=4.0hz,8.0hz),7.33(dd,2h,j=4.0hz,8.0hz),5.11(t,1h,j=8.0hz),3.28(q,2h,j=8.0hz),1.57(m,2h),1.34(m,6h),0.90(t,3h,j=6.76hz)。
[0045]
反应方程式如下:
[0046][0047]
(2)125ml滴液漏斗中将所述中间产物a1(2.76g,10.4mmol,1.0eq)和三乙胺(1.44ml,10.4mmol,4.0eq)溶于干燥thf(60ml)中得到溶液
ⅷ
,氮气氛围下,250ml三口瓶中将实施例1所述化合物ⅰ(1.42g,2.6mmol,4.0eq)溶于干燥thf(60ml)中得到溶液
ⅸ
并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液
ⅷ
,反应14小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:dcm和ch3oh的体积比为50:3),反应结束,冷却至室温后固液分离,固体用thf(10ml
×
3)洗涤后干燥,得到一种三足六脲中性配体(1.9g,产率为75%),记为hu-c6h
13
;1h nmr(400mhz,298k,dmso-d6,ppm):δ=7.89(s,1h),7.77(s,1h),7.52(d,1h,j=7.72hz),7.37(d,1h,j=7.76hz),6.93(m,2h),6.52(m,2h),3.18(m,2h),3.02(q,2h,j=6.48hz),2.60(t,2h,j=6.44hz),1.38(m,2h),1.25(m,6h),0.86(t,3h,j=6.96hz)。
[0048]
反应方程式如下:
[0049][0050]
实施例3
[0051]
一种三足六脲中性配体的制备方法,所述方法步骤如下:
[0052]
(1)125ml滴液漏斗中将十二胺(3.0g,16.2mmol,1.0eq)溶解于干燥thf(60ml)得到溶液ⅵ;氮气氛围下,250ml三口瓶中将4-硝基苯氯甲酸酯(5.0g,24.9mmol,1.5eq)溶于干燥thf(60ml)中得到溶液ⅶ并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液ⅵ,反应14小时后,tlc检测4-硝基苯氯甲酸酯反应完全,生成产物点(比移值rf=0.7,展开剂:dcm),反应结束,体系澄清,冷却至室温,析出大量白色沉淀,过滤除去沉淀,滤液减压浓缩至干,
冷却至室温,加入甲醇(150ml),充分超声搅拌,至沉淀析出完全,过滤沉淀,甲醇(10ml
×
3)洗涤沉淀,干燥,得到中间产物a2(3.6g,产率为64%);1h nmr(400mhz,298k,chcl3,ppm):δ=8.25(dd,2h,j=4.0hz,8.0hz),7.33(dd,2h,j=4.0hz,8.0hz),5.10(t,1h,j=8.0hz),3.28(q,2h,j=8.0hz),1.58(m,2h),1.26(m,18h),0.90(t,3h,j=6.84hz)。
[0053]
反应方程式如下:
[0054][0055]
(2)125ml滴液漏斗中将所述中间产物a2(3.2g,9.1mmol,4.0eq)和三乙胺(1.26ml,9.1mmol,4.0eq)溶于干燥thf(60ml)中得到溶液
ⅷ
,氮气氛围下,250ml三口瓶中将实施例1所述化合物ⅰ(1.2g,2.2mmol,1.0eq)溶于干燥thf(60ml)中得到溶液
ⅸ
并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液
ⅷ
,反应14小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:dcm和ch3oh的体积比为50:3),反应结束,体系澄清;冷却至室温,无沉淀产生,转移至250ml单口瓶旋蒸浓缩至干,加入乙腈(150ml),充分超声搅拌,过滤沉淀物,乙腈洗涤沉淀物(10ml
×
3),沉淀物分散在ch2cl2中,逐滴加入乙醇,加热至沉淀物完全溶解,然后浓缩至有沉淀析出,停止旋蒸,待大量沉淀析出后过滤沉淀,沉淀分别用乙醇、乙醚洗涤,干燥,得到一种三足六脲中性配体(1.2g,产率为48%),记为hu-c
12h25
;1h nmr(400mhz,298k,dmso-d6,ppm):δ=7.86(s,1h),7.76(s,1h),7.51(d,1h,j=7.72hz),7.38(d,1h,j=7.72hz),6.93(m,2h),6.52(m,2h),3.18(m,2h),3.01(q,2h,j=6.6hz),2.60(t,2h,j=5.96hz),1.38(t,2h,j=5.64hz),1.24(m,18h),0.85(t,3h,j=6.48hz)。
[0056]
反应方程式如下:
[0057][0058]
实施例4
[0059]
一种三足六脲中性配体的制备方法,所述方法步骤如下:
[0060]
(1)125ml滴液漏斗中将十八胺(3.4g,12.6mmol,1.0eq)溶解于干燥thf(60ml)得到溶液ⅵ;氮气氛围下,250ml三口瓶中将4-硝基苯氯甲酸酯(3.0g,14.9mmol,1.2eq)溶于干燥thf(60ml)中得到溶液ⅶ并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液ⅵ,反应14小时后,tlc检测4-硝基苯氯甲酸酯反应完全,生成产物点(比移值rf=0.7,展开剂:dcm),反应结束,体系澄清,冷却至室温,析出大量白色沉淀,过滤除去沉淀,滤液减压浓缩至干,冷却至室温,加入甲醇(150ml),充分超声搅拌,至沉淀析出完全,过滤沉淀,甲醇(10ml
×
3)洗涤沉淀,干燥,得到中间产物a3(3.6g,产率为67%);1h nmr(400mhz,298k,chcl3,ppm):δ=8.25(dd,2h,j=4.0hz,8.0hz),7.33(dd,2h,j=4.0hz,8.0hz),5.10(t,1h,j=8.0hz),
3.28(q,2h,j=8.0hz),1.58(m,2h),1.26(m,30h),0.90(t,3h,j=7.0hz)。
[0061]
反应方程式如下:
[0062][0063]
(2)125ml滴液漏斗中将所述中间产物a3(3.6g,8.3mmol,4.0eq)和三乙胺(1.15ml,8.3mmol,4.0eq)溶于干燥thf(60ml)中得到溶液
ⅷ
,氮气氛围下,250ml三口瓶中将实施例1所述化合物ⅰ(1.1g,2.0mmol,1.0eq)溶于干燥thf(60ml)中得到溶液
ⅸ
并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液
ⅷ
,反应14小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:dcm和ch3oh的体积比为50:3),反应结束,体系澄清;冷却至室温,无沉淀产生,转移至250ml单口瓶旋蒸浓缩至干,加入乙腈(150ml),充分超声搅拌,过滤沉淀物,乙腈洗涤沉淀物(10ml
×
3),沉淀物分散在ch2cl2中,充分超声,过滤、干燥,得到一种三足六脲中性配体(2.5g,产率为89%),记为hu-c
18h37
;1h nmr(400mhz,298k,dmso-d6,ppm):δ=7.86(s,1h),7.76(s,1h),7.51(d,1h,j=8.24hz),7.38(d,1h,j=6.8hz),6.93(m,2h),6.50(m,2h),3.18(m,2h),3.01(m,2h),2.60(t,2h,j=5.48hz),1.35(m,2h),1.20(m,30h),0.85(t,3h,j=7.44hz)。
[0064]
反应方程式如下:
[0065][0066]
实施例5
[0067]
一种三足六脲中性配体的制备方法,所述方法步骤如下:
[0068]
(1)125ml滴液漏斗中将烷氧基十二烷基苯胺(2.2g,7.9mmol,1.0eq)溶解于干燥thf(60ml)得到溶液ⅵ;氮气氛围下,250ml三口瓶中将4-硝基苯氯甲酸酯(1.9g,9.4mmol,1.2eq)溶于干燥thf(60ml)中得到溶液ⅶ并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液ⅵ,反应4小时后,tlc检测4-硝基苯氯甲酸酯反应完全,生成产物点(比移值rf=0.6,展开剂:dcm),反应结束,体系澄清,冷却至室温,无沉淀析出,减压浓缩至干,加入甲醇(150ml),充分超声搅拌,至沉淀析出完全,过滤沉淀,甲醇(20ml
×
3)洗涤沉淀,干燥,得到中间产物a4(3.1g,产率为97%);1h nmr(400mhz,298k,chcl3,ppm):δ=8.28(dd,2h,j=2.04hz,7.08hz),7.38(d,2h,j=9.08hz),7.35(d,2h,j=8.44hz),6.90(dd,2h,j=2.12hz,6.84hz),3.94(t,2h,j=6.56hz),1.77(m,2h),1.28(m,18h),0.88(t,3h,j=6.6hz)。
[0069]
反应方程式如下:
[0070][0071]
(2)125ml滴液漏斗中将所述中间产物a4(1.5g,3.4mmol,3.5eq)和三乙胺(0.47ml,3.4mmol,3.5eq)溶于干燥thf(60ml)中得到溶液
ⅷ
,氮气氛围下,250ml三口瓶中将实施例1所述化合物ⅰ(562.5mg,1.0mmol,1.0eq)溶于干燥thf(60ml)中得到溶液
ⅸ
并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液
ⅷ
,反应5小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:dcm和ch3oh的体积比为50:3),反应结束,体系澄清;冷却至室温,无沉淀产生,转移至250ml单口瓶旋蒸浓缩至干,加入乙腈(150ml),充分超声搅拌,过滤沉淀物,乙腈洗涤沉淀物(10ml
×
3),沉淀物分散在ch2cl2中,逐滴加入乙醇,加热至沉淀物完全溶解,然后浓缩至有沉淀析出,停止旋蒸,待大量沉淀析出后过滤沉淀,沉淀分别用乙醇、乙醚洗涤,干燥,得到一种三足六脲中性配体(1.4g,产率为75%),记为hu-phoc
12h25
;1h nmr(400mhz,298k,dmso-d6,ppm):δ=8.91(s,1h),7.97(d,2h,j=9.72hz),7.64(d,1h,j=4.0hz),7.47(d,1h,j=4.0hz),7.36(d,2h,j=4.0hz),7.04(m,2h),6.87(d,2h,j=12.0hz),6.54(t,1h,j=4.0hz),3.92(t,2h,j=8.0hz),3.24(m,2h),2.66(t,2h,j=4.0hz),1.71(m,2h),1.35(m,2h),1.30(m,18h),0.90(t,3h,j=8.0hz)。
[0072]
反应方程式如下:
[0073][0074]
实施例6
[0075]
一种三足六脲中性配体的制备方法,所述方法步骤如下:
[0076]
(1)125ml滴液漏斗中将烷氧基十八烷基苯胺(3g,8.3mmol,1.0eq)溶解于干燥thf(60ml)得到溶液ⅵ;氮气氛围下,250ml三口瓶中将4-硝基苯氯甲酸酯(2g,10mmol,1.2eq)溶于干燥thf(60ml)中得到溶液ⅶ并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液ⅵ,反应10小时后,tlc检测4-硝基苯氯甲酸酯反应完全,生成产物点(比移值rf=0.5,展开剂:dcm),反应结束,体系澄清,冷却至室温,无沉淀析出,减压浓缩至干,加入甲醇(150ml),充分超声搅拌,至沉淀析出完全,过滤沉淀,甲醇(20ml
×
3)洗涤沉淀,干燥,得到中间产物a5(3.7g,产率为85%);1h nmr(400mhz,298k,chcl3,ppm):δ=8.28(dd,2h,j=2.04hz,7.08hz),7.38(d,2h,j=8.44hz),7.35(d,2h,j=8.44hz),6.90(dd,2h,j=2.12hz,6.84hz),3.94(t,2h,j=6.56hz),1.77(m,2h),1.28(m,30h),0.88(t,3h,j=6.6hz)。
[0077]
反应方程式如下:
[0078][0079]
(2)125ml滴液漏斗中将所述中间产物a5(3g,8.3mmol,1.0eq)溶于干燥thf(60ml)中得到溶液
ⅷ
,氮气氛围下,250ml三口瓶中将实施例1所述化合物ⅰ(2g,10mmol,1.2eq)溶于干燥thf(60ml)中得到溶液
ⅸ
并加热至80℃,然后以2秒/滴的速率逐滴滴加溶液
ⅷ
,反应10小时后,tlc检测化合物ⅰ反应完全,生成产物点(比移值rf=0.5,展开剂:dcm和ch3oh的体积比为50:3),反应结束,体系澄清,冷却至室温,无沉淀析出,反应液浓缩至干,加入甲醇(150ml),充分超声搅拌,过滤沉淀物,甲醇洗涤(20ml
×
3),干燥,得到一种三足六脲中性配体(3.7g,产率为85%),记为hu-phoc
18h37
;1h nmr(400mhz,298k,chcl3,ppm):δ=8.28(dd,2h,j=2.04hz,7.08hz),7.38(d,2h,j=8.44hz),7.35(d,2h,j=8.44hz),6.90(dd,2h,j=2.12hz,6.84hz),3.94(t,2h,j=6.56hz),1.77(m,2h),1.28(m,30h),0.88(t,3h,j=6.6hz)。
[0080]
反应方程式如下:
[0081][0082]
实施例7
[0083]
液液萃取硫酸根:
[0084]
将实施例2所述三足六脲中性配体(10mm)与甲基三辛基氯化铵(a336cl)(20mm)溶于chcl3(2ml)中,加入na2so4水溶液(2ml,硫酸根浓度为9.59mm),三足六脲中性配体与硫酸根的摩尔比为1:1;手动上下振动5秒,然后静置5秒。分层结束后取出1ml的水层,通过0.22μm的针头过滤器过滤,然后通过离子色谱仪测定萃取后水溶液中硫酸根的浓度为2.11
±
0.03mm。
[0085]
对实施例3-6中所述三足六脲中性配体进行液液萃取硫酸根,结果与实施例2类似。
[0086]
实施例8
[0087]
竞争条件下液液萃取硫酸根:
[0088]
将实施例2所述三足六脲中性配体(10mm)和a336cl(20mm)溶于chcl3(2ml)中,配置混合盐溶液(na2so4、nano3、kbr和nah2po4各10mm),手动上下振动20秒,然后静置20秒。分层结束后取出1ml的水层,通过0.22μm的针头过滤器过滤,然后用离子色谱仪分析测定萃取
后混合盐溶液中硫酸根的浓度,结果如表1所示。
[0089]
表1
[0090][0091]
对实施例3-6中所述三足六脲中性配体进行竞争条件下液液萃取硫酸根,结果与实施例2类似。
[0092]
实施例9
[0093]
模拟三足六脲中性配体对硫酸根的动态传输效果:
[0094]
u形管中,将实施例2所述三足六脲中性配体(10mm)和a336cl溶于chcl3(3ml)中,三足六脲中性配体与a336cl的摩尔比为1:2;u形管中放置一颗磁子。然后,缓慢地将1ml na2so4溶液(100mm)作为源相加入左侧。在右侧缓慢地加入1ml hcl(600mm)作为接收相;平衡20分钟后,在25
±
2℃下以1000rpm转速搅拌。在实验进行后的6、12、22、34、46和70小时,分别取10μl的源相和接收相,定容至1ml,用离子色谱仪分析测定源相和接收相中硫酸根的浓度,结果如表2所示。
[0095]
表2
[0096][0097][0098]
对比例2
[0099]
u形管中,将对比例1所述三足六脲中性配体(10mm)和a336cl溶于chcl3(3ml)中,三足六脲中性配体与a336cl的摩尔比为1:2;u形管中放置一颗磁子。然后,缓慢地将1ml na2so4溶液(100mm)作为源相加入左侧。在右侧缓慢地加入1ml hcl(600mm)作为接收相;平衡20分钟后,在25
±
2℃下以1000rpm转速搅拌。搅拌30分钟后,配体明显析出粘在u形管管壁上,由此可知,对比例1所述三足六脲中性配体在hcl的作用下溶解变差,从有机溶液中析出来,因此该配体无法用于动态萃取硫酸根。
[0100]
对比例3
[0101]
本对比例中尾端为硝基的三足六脲中性配体按照文献(angew.chem.int.ed.,2011,50(2),486-490)中的方法制备得到,记为hu-ph-no2。
[0102]
u形管中,将hu-ph-no2(10mm)和a336cl溶于chcl3(3ml)中,三足六脲中性配体与a336cl的摩尔比为1:2;u形管中放置一颗磁子。然后,缓慢地将1ml na2so4溶液(100mm)作为源相加入左侧。在右侧缓慢地加入1ml hcl(600mm)作为接收相;平衡20分钟后,在25
±
2℃下以1000rpm转速搅拌。搅拌30分钟后,配体明显析出粘在u形管管壁上,由此可知,尾端为硝基的三足六脲中性配体在hcl的作用下溶解变差,从有机溶液中析出来,因此该配体无法用于动态萃取硫酸根。
[0103]
对比例4
[0104]
本对比例中尾端为硝基的三足六脲中性配体按照文献(angew.chem.int.ed.,2011,50(2),486-490)中的方法制备得到,记为hu-ph-no2。
[0105]
将hu-ph-no2(12mm)和a336cl溶于chcl3(2ml)中,加入na2so4水溶液(2ml,硫酸根浓度为10mm),室温下剧烈上下摇晃10秒,离心后取出上层水相(1ml),用0.22μm的针头过滤器过滤,然后用离子色谱仪分析测定硫酸根浓度,结果如表3所示,由此得到硫酸根的萃取效率结果如图1所示。
[0106]
然后轻轻取出水相,加入hcl(400mm,2ml)进行反萃取,之后取出上层水相(1ml),用0.22μm的针头过滤器过滤,然后用离子色谱仪分析测定硫酸根浓度,结果如表3所示,由此得到硫酸根的反取效率结果如图1所示。
[0107]
对比例5
[0108]
将对比例4中的三足六脲中性配体替换为对比例1所述三足六脲中性配体(13mm),其余同对比例4。
[0109]
硫酸根萃取和反萃取后的浓度结果如表3所示,萃取和反萃取效率如图2所示。
[0110]
实施例10
[0111]
将对比例4中的三足六脲中性配体替换为实施例2所述三足六脲中性配体(15mm),其余同对比例4。
[0112]
硫酸根萃取和反萃取后的浓度结果如表3所示,萃取和反萃取效率如图3所示。
[0113]
实施例11
[0114]
将对比例4中的三足六脲中性配体替换为实施例5所述三足六脲中性配体(13mm),其余同对比例4。
[0115]
硫酸根萃取和反萃取后的浓度结果如表3所示,萃取和反萃取效率如图4所示。
[0116]
表3
[0117][0118]
由表3的结果可知,与hu-ph-no2和hu-phch3相比,hu-c6h
13
和hu-phoc
12h25
在重复使用8次之后,仍能保持80%以上的萃取效率,表明本发明所述三足六脲中性配体具有较好的重复使用性能。
[0119]
综上所述,发明包括但不限于以上实施例,凡是在本发明的精神和原则之下进行的任何等同替换或局部改进,都将视为在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1