一种胆固醇衍生物及其制备方法和应用
本发明属于药物化学领域,具体而言,涉及一种三环类fxr受体拮抗剂,其制备方法以及该类fxr拮抗剂在fxr拮抗活性方面的用途。
背景技术:
1、胆汁酸除了促进维生素和脂质吸收等生理功能外,还能作为信号分子参与到全身的代谢调控中。胆汁酸作为信号分子可以结合膜受体和核受体两类,而核受体中法尼酯衍生物受体x(fxr)是专一性被胆汁酸识别的受体。fxr受体在维持胆汁酸稳态方面起着非常重要的作用。一方面,肝脏中fxr受体被激活后,可以诱导小异源二聚体伴侣(shp)的表达,抑制胆汁酸合成途径中的限速酶cyp7a1和cyp8b1的表达,从而降低胆汁酸的合成。而在肠道上皮细胞中,fxr受体的激活可以诱导成纤维细胞生长因子19(fgf19,在鼠中为fgf15)的表达,fgf19/15结合肝脏中的fgfr4受体,抑制cyp7a1的表达而降低胆汁酸的合成。除此之外,fxr受体还与糖脂代谢密切相关,另外,研究表明fxr受体与胆汁淤积、代谢紊乱等多种疾病密切相关。
2、目前,多项研究表明fxr拮抗剂可以改善小鼠的代谢。例如,研究人员发现二甲双胍可以改变人肠道菌群的组成,降低胆盐水解酶的活性,从而使得有fxr拮抗活性的结合型胆汁酸gudca和tudca的含量升高,进而抑制肠道fxr的表达,改善代谢包括高血糖。小鼠经过抗氧化剂tempol处理后,也会引起肠道内菌群的改变,降低胆盐水解酶的活性,肠道内结合型胆汁酸t-β-mca增加,从而抑制fxr信号转导。肠道fxr信号的抑制引起线粒体功能的改善和神经酰胺合成的抑制,导致血清神经酰胺水平降低。减少的循环神经酰胺会下调肝脏srebp1c和cidea的表达,从而降低肝脏脂肪变性。天然fxr拮抗剂isodca可以通过拮抗树突状细胞中的fxr受体来减弱其免疫刺激,增加foxp3的诱导作用,增加了肠道cd4+调节性t细胞的数量,进而影响肠道健康。因此,fxr拮抗剂为一些代谢性疾病的治疗提供了新的思路。
3、目前报道的fxr拮抗剂可以分为天然产物类和小分子类。天然产物类fxr拮抗剂最多的是一些结合型的胆汁酸,包括t-β-mca,gly-mac,gudca,tudca等。天然产物没药甾酮(guggulaterone,gs)是首个被报道的fxr受体拮抗剂,但是作为一种胆汁酸激活受体的拮抗剂,没药甾酮对于fxr并没有选择性。在拮抗试验中,gs能够降低cdca的fxr激动活性,ic50为25μm。对其他核受体的ic50值在0.32-62μm之间。flg249是首个被报道的作用于小鼠回肠fxr下游的非甾体小分子类fxr受体拮抗剂。口服flg249能够下调小鼠回肠中的fxr靶基因fgf15,asbt和shp的mrna水平,并且组织分布研究表明flg主要集中在回肠中。
4、
5、目前存在的fxr拮抗剂天然来源有限,且合成难度较大,针对此类化合物的结构改造较少,尚需研发。
技术实现思路
1、本发明的目的在于提供一种用作法尼酯衍生物x受体(fxr)拮抗剂的化合物。
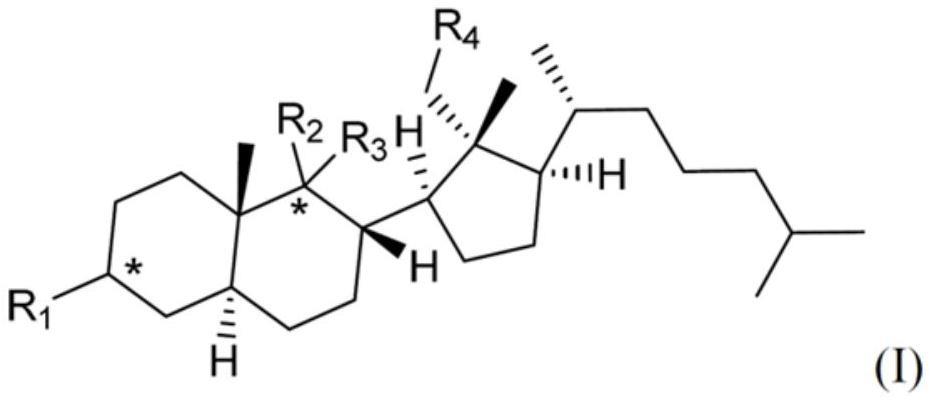
2、本发明的第一方面,提供一种通式(i)所示的化合物,或其药学上可接受的盐,
3、
4、式中,
5、r1选自:氢、羟基、卤素、c1-c6烷氧基、3至10元环烷氧基、=o、=n-oh、rd-c(=o)-o-;其中rd为取代或未取代的以下基团:c1-c8烷基、c3-c10环烷基、c6-c10芳基、5-7元杂芳基、ranh-或rao-;其中,各ra独立选自:c1-c8烷基、c3-c10环烷基、c6-c10芳基、5-7元杂芳基;
6、r2为氢、羟基、卤素或c1-c6烷基;r3选自:氢、羟基、卤素、c1-c6烷氧基、3至10元环烷氧基、re-c(=o)-o-或re-oc(=o)-;其中各re独立地为氢或取代或未取代的以下基团:c1-c8烷基、c3-c10环烷基、c6-c10芳基、5-7元杂芳基、rbnh-或rbo-;其中,各rb独立选自:c1-c8烷基、c3-c10环烷基、c6-c10芳基、5-7元杂芳基;
7、或者r2与r3与连接的碳形成c=o或c=n-oh;
8、r4选自:氢、羟基、羟甲基、甲酰基、-(c2-c6亚烯基)c(=o)-rf-rg、-(c1-c6亚烷基)c(=o)-rf-rg,其中,x为-nh-、-o-、-s-或-nhso2-;rc为氢、未取代或取代的c1-c6烷基、未取代或取代的c6-c10芳基;rf为o、s或nh;rg为氢、取代或未取代的c1-c6烷基;
9、各*独立地表示r构型、s构型或消旋;
10、所述取代是指被基团上的氢被选自下组的一个或多个取代基取代:羟基、卤素、c1-c6烷基、c1-c6烷氧基、羧基(-cooh)、磺酸基(-so2oh)。
11、在另一优选例中,r1为羟基或rd-c(=o)-o-;其中rd为取代或未取代的以下基团:c1-c6烷基、c3-c8环烷基、c6-c10芳基、5-7元杂芳基、ranh-或rao-;其中,各ra独立选自为c1-c6烷基;
12、所述取代是指被基团上的氢被选自下组的一个或多个取代基取代:卤素、c1-c6烷、c1-c6烷氧基。
13、在另一优选例中,r1为oh或-coo(c1-c4烷基)。
14、在另一优选例中,与r1相连的c的构型为s型或r型。
15、在另一优选例中,r2为氢;r3为羟基、卤素、c1-c4烷氧基、3至8元环烷氧基、re-c(=o)-o-或re-oc(=o)-;其中各re独立地为氢或取代或未取代的以下基团:c1-c6烷基、c3-c8环烷基、c6-c10芳基、5-7元杂芳基、rbnh-或rbo-;其中,各rb独立选自:c1-c6烷基、c3-c8环烷基、c6-c10芳基、5-7元杂芳基;所述取代是指被基团上的氢被选自下组的一个或多个取代基取代:羟基、卤素、c1-c4烷基、c1-c4烷氧基、羧基;
16、或者r2与r3与连接的碳形成c=o。
17、在另一优选例中,r2为氢;r3为羟基。在另一优选例中,与r2、r3相连的c的构型为s型或r型。在另一优选例中,r2与r3与连接的碳形成c=o。
18、在另一优选例中,r4选自:氢、羟基、羟甲基、甲酰基、-(c2-c4亚烯基)c(=o)-rf-rg、-(c1-c4亚烷基)c(=o)-rf-rg,其中,x为-nh-、-o-、-s-或-nhso2-;rc为氢、未取代或取代的c1-c4烷基、未取代或取代的苯基;rf为o、s或nh;rg为氢、取代或未取代的c1-c4烷基;
19、所述取代是指被基团上的氢被选自下组的一个或多个取代基取代:c1-c4烷基、羧基(-cooh)、磺酸基(-so2oh)。
20、在另一优选例中,r4选自:-coorc、-(c2-c4亚烯基)cooh、-(c1-c4亚烷基)cooh、-conhso2rc、-conhrc;rc为氢、未取代或取代的c1-c4烷基、未取代或取代的苯基;所述取代是指被基团上的氢被选自下组的1、2或3个取代基取代:甲基、乙基、正丙基、异丙基、羧基(-cooh)、磺酸基(-so2oh)。
21、在另一优选例中,所述化合物选自下组:
22、
23、
24、本发明的第二方面,提供一种第一方面所述的化合物或其药学上可接受的盐的制备方法,所述方法以胆固醇为原料,c3位的羟基可以改变构型;通过在化学反应将双键移c9和c10上,再通过氧化开环得到含有醛基侧链的化合物,然后醛基经过氧化得到羧基,羧基上可进行一系列的取代;或者醛基可以通过延长碳链的方式得到碳链增加的羧基;此外,c9位上的羰基也可进行还原得到不同构型羟基取代的第一方面所述的化合物,各取代基的定义如第一方面所述。
25、本发明所述的化合物,结构如式h3、h4、h5,h6或h7所示,通过以下路线制备:
26、路线1:
27、
28、(i)式h1化合物经过臭氧化得到式h2化合物;
29、(ii)式h2化合物经过氧化醛基得到式h3化合物;
30、任选地(iii)式h3化合物与氨基酸或磺酰胺反应得到式h4化合物;
31、路线2:式h3化合物与硼氢化钠反应得到式h5化合物;
32、
33、或路线3:
34、
35、(i’)式h2化合物与丙二酸,吡啶和哌啶反应得到式h6化合物;
36、任选地(ii’)式h6化合物与硼氢化钠反应得到式h7化合物,
37、各式中,r5为oh或-coo(c1-c4烷基),与r5相连的c的构型为r或者s型,或者r或者s构型的乙酰基;
38、a为-nh-或-nhso2-;
39、r6选自:氢、未取代或取代的c1-c6烷基、未取代或取代的c6-c10芳基;所述取代是指被基团上的氢被选自下组的一个或多个取代基取代:羟基、卤素、c1-c6烷基、c1-c6烷氧基、羧基(-cooh)、磺酸基(-so2oh);
40、r7为氢或羟基;
41、r8为氢或羟基;
42、r9为氢或羟基;
43、r10为氢或羟基。
44、本发明的第三方面,提供一种药物组合物,包含:
45、第一方面所述的通式(i)所示的化合物或其药学上可接受的盐;和
46、药学上可接受的载体。
47、“药学上可接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的活性成分(通式(i)所示的化合物或其药学上可接受的盐)以及它们之间相互掺和,而不明显降低活性成分的药效。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
48、本发明的活性成分或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、直肠、肠胃外(静脉内、肌肉内或皮下)等。
49、用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。
50、用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性成分外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、矫味剂和香料。
51、除了活性成分外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。
52、用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。
53、本发明化合物可以单独给药,或者与其他治疗药物联合给药。
54、使用药物组合物时,是将安全有效量的本发明化合物适用于需要治疗的哺乳动物(如人),其中施用时剂量为药学上认为的有效给药剂量,对于60kg体重的人而言,日给药剂量通常为1~2000mg,优选20~500mg。当然,具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。
55、本发明的第四方面,提供第一方面所述的通式(i)所示的化合物、或其药学上可接受的盐或第三方面所述的药物组合物用途,(i)用于制备法尼酯衍生物x受体(fxr)拮抗剂;(ii)用于制备治疗与法尼酯衍生物x受体相关的疾病的药物;或(iii)用于制备治疗代谢性疾病的药物。
56、在另一优选例中,所述与法尼酯衍生物x受体相关的疾病选自:高血脂、胆汁酸淤积、糖尿病(如2型糖尿病)、肥胖、非酒精性脂肪肝、胆汁性肝硬化、高胆固醇血症。
57、在另一优选例中,所述代谢性疾病选自:高血脂、胆汁酸淤积、糖尿病、肥胖、非酒精性脂肪肝、胆汁性肝硬化、高胆固醇血症。
58、本发明的化合物,能有效拮抗fxr受体,可在微摩尔浓度下拮抗fxr受体,与现有的一些天然来源的fxr拮抗剂相比,具有更丰富的天然来源和更简便的合成方法。
59、体外活性测试证明,该类化合物具有良好的fxr拮抗活性,并且在体内可以调控fxr受体下游基因的表达。因此该类化合物可以作为治疗代谢性疾病的候选药物。
60、应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。说明书中所揭示的各个特征,可以被任何提供相同、均等或相似目的的替代性特征取代。限于篇幅,在此不再一一累述。
- 还没有人留言评论。精彩留言会获得点赞!