一种环戊烯并喹嗪并咪唑类化合物及其制备方法和应用
1.本发明属于化学药技术领域,具体涉及一种环戊烯并喹嗪并咪唑类化合物及其制备方法和应用。
背景技术:
2.结肠癌是一种发生在人体结肠部位的消化道恶性肿瘤,是常见疾病,常发生在直肠和乙状结肠交界的部位,慢性结肠炎患者、结肠息肉患者、男性肥胖者等为易感人群。
3.外科手术是治疗结肠癌的首选方法,但因留下残存病灶,导致易复发和转移;放疗和化疗也是治疗结肠癌的常见方法,但放疗不仅有极大的副作用,会造成患者身体衰弱、免疫功能下降、骨髓抑制、消化道障碍等严重不良反应,还会损害患者肝肾等主要脏器;化疗也会引起很多毒副反应,容易引起合并症、后遗症等。因此,获得更多能够用于结肠癌的化合物对于结肠癌的药物开发和临床治疗都具有重要意义。
技术实现要素:
4.针对以上问题,本发明提供一种环戊烯并喹嗪并咪唑类化合物及其制备方法和应用,该化合物能够抑制结肠癌细胞增殖,可用于制备抗肿瘤药物。
5.为达到上述发明目的,本发明实施例采用了如下技术方案:
6.第一方面,本发明实施例提供了一种环戊烯并喹嗪并咪唑类化合物,其结构式如式i所示:
[0007][0008]
经实验验证,该化合物对于结肠癌细胞的增殖具有显著的抑制作用。目前尚未发现该环戊烯并喹嗪并咪唑类化合物的相关报道。
[0009]
以及,本发明还提供上述环戊烯并喹嗪并咪唑类化合物的制备方法,包括以下步骤:
[0010]
步骤a、将京尼平与组氨酸溶于ph为4~10的非含氮类缓冲溶液中,在30~80℃反应2h以上,得反应液;
[0011]
步骤b、将所述反应液加至小孔树脂层析柱,依次采用体积比为0:100、5:95、1:9、2:8、4:6的甲醇
–
水溶剂进行梯度洗脱,收集体积比为4:6的甲醇
–
水溶剂洗脱所得洗脱液,
即得所述环戊烯并喹嗪并咪唑类化合物。
[0012]
本发明利用京尼平与组氨酸在特定溶液中、在特定温度下反应,获得含有该环戊烯并喹嗪并咪唑类化合物的反应液,继而通过小孔树脂柱色谱,可将该环戊烯并喹嗪并咪唑类化合物与反应液中的其它化合物进行分离,即可在洗脱液中获得该环戊烯并喹嗪并咪唑类化合物。
[0013]
结合第一方面,该制备方法还可包括将所述洗脱液用制备液相色谱分离,根据所述环戊烯并喹嗪并咪唑类化合物色谱峰的出峰时间收集洗脱流分,浓缩、干燥,即可在特定洗脱流分中得到该环戊烯并喹嗪并咪唑类化合物的纯品。
[0014]
优选地,所述制备液相色谱的色谱条件为:色谱柱为碳十八键合硅胶色谱柱,流动相为体积比为(80:20)~(60:40)的0.05%~0.20%(v/v)甲酸水
–
甲醇,等度洗脱。
[0015]
在该色谱条件下,色谱柱可选zorbax prephtxdb c18,规格为21.2
×
250mm,7μm;流动相可选体积比为65:35的0.1%(v/v)甲酸水
–
甲醇;等度洗脱的流速可选10ml/min;进样量可选1ml。
[0016]
结合第一方面,所述缓冲溶液为磷酸二氢钾-氢氧化钠缓冲溶液、pbs缓冲溶液、醋酸钠-冰乙酸或硼砂-碳酸钠缓冲溶液。
[0017]
结合第一方面,所述缓冲溶液的ph优选为7.35。
[0018]
结合第一方面,步骤a中所述反应的温度优选32~42℃。
[0019]
结合第一方面,步骤b中每种洗脱溶剂的用量可选3个柱体积,以确保目标化合物与其它化合物的分离,并使目标化合物得到更好的富集。
[0020]
第二方面,本发明实施例还提供了上述环戊烯并喹嗪并咪唑类化合物在制备抗肿瘤药物中的应用。
[0021]
通过实验验证发现,该化合物能明显抑制人结肠癌细胞系sw480细胞、小鼠结肠癌细胞系ct26细胞的增殖活性(p《0.05),故可应用于制备抗肿瘤药物。
[0022]
结合第二方面,所述抗肿瘤药物为抗结肠癌药物。
[0023]
结合第二方面,所述抗肿瘤药物为抗结肠癌的靶向制剂。将该化合物制成靶向制剂,能够提高药效,减少对其它组织的影响。
附图说明
[0024]
图1为本发明实施例1中genihistidine b的1h-nmr谱图;
[0025]
图2为本发明实施例1中genihistidine b的
13
c-nmr谱图;
[0026]
图3为本发明实施例1中genihistidine b的1h-1
h cosy谱图;
[0027]
图4为本发明实施例1中genihistidine b的hsqc谱图;
[0028]
图5为本发明实施例1中genihistidine b的hmbc谱图;
[0029]
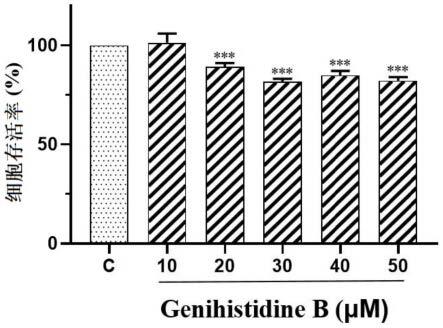
图6为本发明实施例8中genihistidine b的体外抑制人结直肠癌细胞系sw480细胞增殖活性
[0030]
图7为本发明实施例8中genihistidine b的体外抑制小鼠结直肠癌细胞系ct26细胞增殖活性。
具体实施方式
[0031]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用于解释本发明,并不用于限定本发明。
[0032]
结肠癌发病率较高,中晚期的治愈率和生存率下降,为获得更多可用于结肠癌治疗的化合物,本发明实施例提供了一种环戊烯并喹嗪并咪唑类化合物(命名为genihistidine b),其结构式如式i所示:
[0033][0034]
实验发现,genihistidine b能够抑制人结肠癌细胞系sw480细胞、小鼠结肠癌细胞系ct26细胞的增殖活性(p《0.05),对于结肠癌的治疗具有重要意义。
[0035]
本发明实施例还提供了该化合物的制备方法,包括以下步骤:
[0036]
步骤a、将京尼平与组氨酸溶于ph为4~10的非含氮类缓冲溶液中,在30~80℃反应2h以上,得反应液;
[0037]
步骤b、将所述反应液加至小孔树脂层析柱,依次采用体积比为0:100、5:95、1:9、2:8、4:6的甲醇
–
水溶剂进行梯度洗脱,收集体积比为4:6的甲醇
–
水溶剂洗脱所得洗脱液,即可在洗脱液中得到genihistidine b。
[0038]
在该制备方法中,步骤b得到的洗脱液还可经制备液相色谱分离进一步纯化,以获得genihistidine b的纯品。纯化方法可选择以下操作:将步骤b所得洗脱液用制备液相色谱分离,色谱柱为碳十八键合硅胶色谱柱,流动相为体积比为(80:20)~(60:40)的0.05%~0.20%(v/v)甲酸水
–
甲醇,等度洗脱。其中,色谱柱可选zorbax prephtxdb c18,规格为21.2
×
250mm,7μm;流动相可选体积比为65:35的0.1%(v/v)甲酸水
–
甲醇;等度洗脱的流速可选10ml/min。在该条件下,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩、干燥,即可得到genihistidine b的纯品。
[0039]
在该制备方法中,本发明对京尼平与组氨酸的质量比不做限定,二者在任何质量比下均可通过上述制备方法制得该环戊烯并喹嗪并咪唑类化合物。但需要说明的是,该制备方法不对原料质量比进行限定并不代表京尼平与组氨酸在任何比例下均可全部参与反应,投料量多的原料会在反应过程中被剩余。为了确保产物的收率,减少原料浪费,京尼平与组氨酸的质量比优选采用1:3~4,更优选的质量比为1:3.4~3.5。
[0040]
本发明对缓冲溶液的类型不做限定,使用非含氮类常用缓冲溶液即可,例如磷酸二氢钾
–
氢氧化钠缓冲溶液、pbs缓冲溶液、醋酸钠-冰乙酸或硼砂-碳酸钠缓冲溶液。
[0041]
缓冲溶液的ph优选采用7.35。在其它条件相同的情况下,当ph为7.35时,该环戊烯并喹嗪并咪唑类化合物的产量最高。
[0042]
该制备方法的反应温度优选为32~42℃,反应时间为60h。在优选的原料质量比、反应温度、ph值和反应时间下,能够生成更多的目标产物genihistidine b。
[0043]
在小孔树脂柱色谱洗脱过程中,每种所述洗脱溶剂的用量均为3个柱体积,以提高genihistidine b与其它化合物的分离效果,并使genihistidine b得以富集。小孔树脂层析柱可选用chp20/p120小孔树脂作为填料。
[0044]
以下通过具体实施例来对本发明的方案进行说明。
[0045]
实施例1
[0046]
本实施例提供了一种环戊烯并喹嗪并咪唑类化合物genihistidine b及其制备方法。
[0047]
genihistidine b的制备方法具体包括以下步骤:
[0048]
步骤a、取京尼平113mg、组氨酸387.5mg,精密称定,分别置于100ml量瓶中,加ph=7.35的pbs缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和25mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=7.35的pbs缓冲溶液至450ml,混匀,于37℃水浴反应60h后取出,即得反应液;
[0049]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0050]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.1%(v/v)甲酸水
–
甲醇(65:35),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为4.3%(产率=genihistidine b的重量
÷
京尼平与组氨酸的总重量
×
100%,下同),纯度为99.2%(纯度=从制备液相中收集的流分在超高效液相色谱仪中进行检测,得到的genihistidine b的峰面积
÷
所有超高效液相色谱中的峰面积之和
×
100%,下同)。
[0051]
采用nmr(1h-nmr、
13
c-nmr、1h-1
h cosy、hsqc、hmbc)技术表征genihistidine b的化学结构,1h-nmr谱图如图1所示,
13
c-nmr谱图如图2所示,1h-1
h cosy谱图如图3所示,hsqc谱图如图4所示,hmbc谱图如图5所示。
[0052]
在1h-nmr谱中,显示特征质子信号δ7.69(1h,s,h-2),三个亚甲基质子信号δ2.75、2.16(h-5),δ3.19、2.99(h-15),δ4.24(h-19)。高场显示1个甲氧基信号δ3.67(h-18)。
13
c-nmr谱中显示17个碳信号,主要包括1个酯羰基信号δ171.7,4个芳香碳信号δ149.3、99.5、131.8、144.6,2个亚甲基信号δ38.6、59.8,3个次甲基信号δ37.9、46.9、51.5,此外还显示一个甲氧基信号δ51.1;δ177.3信号推测为羧基碳信号,δ130.6、134.6、124.5为咪唑环上的碳信号。化合物1h-1
h cosy谱中,显示h-6和h-5、h-4和h-8、h-16和h-15之间存在相关;在hmbc谱中,观察到h-9与c-14信号相关,h-16与c-9信号相关,可确定京尼平与组氨酸通过c-9、c-10位相连接;此外还观察到h-2与c-16、h-16与c-9存在相关。
[0053]
经上述nmr谱图可知,该化合物的结构式如式i所示:
[0054][0055]
实施例2
[0056]
本实施例提供了一种genihistidine b的制备方法。
[0057]
具体包括以下步骤:
[0058]
步骤a、取京尼平113mg、组氨酸339mg,精密称定,分别置于100ml量瓶中,加ph=7.35的pbs缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和22mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=7.35的pbs缓冲溶液至450ml,混匀,于32℃水浴反应55h后取出,即得反应液;
[0059]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0060]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.2%(v/v)甲酸水
–
甲醇(60:40),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集11~12min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为38%,纯度为90.4%。
[0061]
实施例3
[0062]
本实施例提供了一种genihistidine b的制备方法。
[0063]
具体包括以下步骤:
[0064]
步骤a、取京尼平113mg、组氨酸452mg,精密称定,分别置于100ml量瓶中,加ph=7.35的pbs缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和29mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=7.35的pbs缓冲溶液至450ml,混匀,于42℃水浴反应65h后取出,即得反应液;
[0065]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0066]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.05%(v/v)甲酸水
–
甲醇(80:20),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集14~16min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为3.8%,纯度为99.0%。
[0067]
实施例4
[0068]
本实施例提供了一种环戊烯并喹嗪并咪唑类化合物genihistidine b及其制备方
法。
[0069]
genihistidine b的制备方法具体包括以下步骤:
[0070]
步骤a、取京尼平113mg、组氨酸387.5mg,精密称定,分别置于100ml量瓶中,加ph=9的硼砂-碳酸钠缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和25mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=9的硼砂-碳酸钠缓冲溶液至450ml,混匀,于37℃水浴反应60h后取出,即得反应液;
[0071]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0072]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.1%(v/v)甲酸水-甲醇(65:35),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为4.0%,纯度为98.9%。
[0073]
实施例5
[0074]
本实施例提供了一种环戊烯并喹嗪并咪唑类化合物genihistidine b及其制备方法。
[0075]
genihistidine b的制备方法具体包括以下步骤:
[0076]
步骤a、取京尼平113mg、组氨酸387.5mg,精密称定,分别置于100ml量瓶中,加ph=6的醋酸钠-冰乙酸缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和25mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=6的醋酸钠-冰乙酸缓冲溶液至450ml,混匀,于37℃水浴反应60h后取出,即得反应液;
[0077]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0078]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.1%(v/v)甲酸水-甲醇(65:35),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为2.5%,纯度为98.3%。
[0079]
实施例6
[0080]
本实施例提供了一种环戊烯并喹嗪并咪唑类化合物genihistidine b及其制备方法。
[0081]
genihistidine b的制备方法具体包括以下步骤:
[0082]
步骤a、取京尼平113mg、组氨酸387.5mg,精密称定,分别置于100ml量瓶中,加ph=10的硼砂-碳酸钠缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和25mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=10的硼砂-碳酸钠缓冲溶液至450ml,混匀,于37℃水浴反应60h后取出,即得反应液;
[0083]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0084]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.1%(v/v)甲酸水-甲醇(65:35),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为1.2%,纯度为97.3%。
[0085]
实施例7
[0086]
本实施例提供了一种环戊烯并喹嗪并咪唑类化合物genihistidine b及其制备方法。
[0087]
genihistidine b的制备方法具体包括以下步骤:
[0088]
步骤a、取京尼平113mg、组氨酸387.5mg,精密称定,分别置于100ml量瓶中,加ph=4的醋酸钠-冰乙酸缓冲溶液溶解并定容至刻度,混匀,即得浓度分别为5和25mmol/l的储备液。分别移取上述储备液各90ml,置于烧杯中,加ph=4的醋酸钠-冰乙酸缓冲溶液至450ml,混匀,于37℃水浴反应60h后取出,即得反应液;
[0089]
步骤b、将反应液采用chp20/p120小孔树脂通过柱色谱法进行分离,依次用甲醇:水(0:100、5:95、1:9、2:8、4:6)溶剂系统梯度洗脱(每个洗脱系统均洗脱3个柱体积),得到一系列洗脱液;合并40%甲醇水洗脱液(即甲醇:水为4:6),浓缩,供制备液相分离纯化用;
[0090]
步骤c、取40%甲醇水洗脱液的浓缩液,采用制备液相色谱分离纯化,色谱柱为zorbax prephtxdb c18(21.2
×
250mm,7μm),流动相为0.1%(v/v)甲酸水-甲醇(65:35),以10ml/min的流速等度洗脱,检测波长为254nm,进样量1ml,根据出峰时间收集12.0~13.0min的洗脱流分,浓缩,干燥,即得genihistidine b,产率为1.9%,纯度为98.6%。
[0091]
实施例8
[0092]
3-(4,5-二甲基-2-噻唑)-2,5二苯基四氮唑溴盐(mtt)可被活细胞线粒体内的脱氢酶还原生成深紫色结晶状产物甲瓒。细胞增殖越快,脱氢酶活性越高,形成的甲瓒越多。甲瓒可以被dmso完全溶解成蓝色,在570nm波长处有强吸收峰,通过酶标仪检测570nm波长的吸光度值,即可评价活细胞数和增殖活性。本实施例利用mtt比色法测定细胞活力来验证genihistidine b抑制结肠癌细胞增殖活性的效果。
[0093]
1、试剂和仪器、耗材
[0094]
(1)试剂:mtt,solarbio;dmso,solarbio;pbs缓冲溶液,佰思诺(天津)生物科技有限公司。
[0095]
(2)仪器、耗材:450型酶标仪,上海伯乐公司;96孔板,nest。
[0096]
2、实验方法
[0097]
mtt工作溶液的配制:将25mg mtt粉末加入到5ml pbs缓冲溶液中,配制成5mg/ml的溶液,分装后置-20℃避光保存。
[0098]
选取处于对数生长期的结肠癌细胞系sw480,用0.25%(g/ml)的胰蛋白酶将细胞团消化成单个细胞,加入三倍体积的dmem完全培养基终止消化,1000rpm离心5min,弃去上清液。加入5ml的dmem完全培养基,混合均匀后吸取10μl的细胞悬液,沿盖玻片边缘将细胞液注入计数板中对细胞进行计数。采用dmem完全培养基制备4
×
104个/ml的细胞混悬液,接种于96孔板中,每孔200μl,并置于co2培养箱中培养24h。移除96孔板中培养液,分别加入用dmem完全培养基稀释的浓度分别为10、20、30、40、50μm的genihistidine b溶液,每个浓度设置10个复孔;同时,设立空白对照组。继续置于co2培养箱中培养24h后移除测试药物,加
入100μl/孔mtt溶液(用dmem完全培养基溶解,浓度为0.5mg/ml),37℃孵育4h后显微镜观察可见结晶状的深紫色产物,加入100μl/孔dmso,溶解深紫色结晶,在570nm波长下检测吸光度值(a)。
[0099]
采用相同方法处理结肠癌细胞系ct26,并采用rpmi 1640完全培养基制备4
×
104个/ml的细胞混悬液,接种于96孔板中,每孔200μl,并置于co2培养箱中培养24h。移除96孔板中培养液,加入rpmi 1640完全培养基稀释的浓度梯度分别为5、10、15、20、25、30μm的genihistidine b溶液,每个浓度设置10个复孔;同时,设立空白对照组。继续置于co2培养箱中培养24h后移除测试药物,加入100μl/孔mtt溶液(rpmi 1640完全培养基溶解,浓度为0.5mg/ml),37℃孵育4h后显微镜观察可见结晶状的深紫色产物,加入100μl/孔dmso,溶解深紫色结晶,在570nm波长下检测吸光度值(a)。
[0100]
以下式计算结肠癌细胞的细胞存活率:
[0101]
细胞存活率/%=a
测试药物
/a
空白
×
100%
[0102]
3、统计学分析
[0103]
研究数据均采用graphpad prism 8统计软件进行分析。空白对照组与实验组间差异分析采用单因素方差分析(one-way anova)。
[0104]
4、实验结果
[0105]
结果显示:与空白对照组相比,genihistidine b明显抑制人结肠癌细胞系sw480细胞(p《0.001)、小鼠结肠癌细胞系ct26细胞的增殖活性(p《0.001),且存在一定剂量依赖关系,结果如图6、图7所示。图中c为空白对照组;***表示p《0.001。
[0106]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1