一种基于芴单元的联咔唑材料及其制备方法和应用
1.本发明属于有机电致发光材料技术领域,涉及基于芴单元修饰的深蓝光联咔唑材料的及其制备方法和应用。
技术背景
2.有机发光二极管(oled)是1987年由tang和vanslyke(appl.phys.lett.1987,51,913.doi.org/10.1063/1.98799)这两位科学家首次提出的,它作为新一代全彩色显示器以及在固体照明光源材料上的广泛应用而在科学界受到了研究者们极大的关注。三十年以来,oled已经逐渐成为可商业化的显示材料广泛应用于手机、电脑等电子设备,尽管在该领域取得了重大进展,但想要真正实现全彩色显示,就需要实现色纯度很高的红色、绿色和蓝色发射。目前来说,蓝色磷光oled由于寿命短、效率低等缺点无法满足业界的需求(j.am.chem.soc.,2011,133(44):17895.doi.org/10.1021/ja207554h),开发出成本低廉、价格便宜的高效稳定蓝色和深蓝色oled材料依旧是一个巨大的挑战。联咔唑是一种结构稳定的富电子体,作为双极传输中心其同时具备电子传输和电荷传输的能力,已有多种以联咔唑为中心的衍生物可用于蓝色磷光oled的主体材料(acs appl.mater.interfaces 2014,6,(17),14874.doi.org/10.1021/am502848c),但其作为磷光发光的主体材料蓝光发射纯度以及热稳定性均有待提高,且电子传输的能力稍显不足。
技术实现要素:
3.鉴于上述不足,本发明通过引入缺电子基团,构建给受体结构的设计思路,既可以有效提高其三线态能级,同时材料也具备优异的热稳定性能。芴基的引入能够调控材料的发射波长向更深蓝光区域移动,合成了一种具有双极传输中心的蓝色发光主体材料。
4.本发明提出一种条件温和、成本低廉的合成方法获取具有双极传输性能的材料,通过将拟卤素氰基和芴单元引入到联咔唑的中心主体上,合成了一种给受体结构dfczphcn。拟卤素取代基效应能够促进系间窜越的过程,并且淬灭咔唑基元的激子,在聚集态下,氰基苯和咔唑之间所形成的电荷转移态可作为单线态和三线态之间的过渡态,进而促进激子的系间窜越和反系间窜越过程。相比于单体咔唑结构,二联咔唑的中心结构也可以进行空穴传输,尽管三线态能级有所降低,但是物质的热稳定性和玻璃化转变温度得到了极大的提高,成膜性能更好。烷基链的引入可以提高化合物的成膜性能,可有效抑制组分的聚集。芴作为蓝光材料的重要结构片段,它的引入一方面促进深蓝色发光,同时保证了良好的双极性和较高的三重态能级。实现了调控材料的发射光谱的红移,实现了422nm处的蓝色发光,可用于高纯度的蓝光oled器件。
5.第一方面,本发明提供一种基于芴单元的联咔唑材料,是一种含芴单元和氰基修饰的双极性联咔唑材料,以下简称dfczphcn,其化合物的分子结构如下:
[0006][0007]
第二方面,本发明还提供上述化合物dfczphcn的具体制备方法,合成路线如下:
[0008][0009][0010]
与上述合成路线对应的,具体制备步骤如下:
[0011]
步骤(i)具体为,对溴苯酚在无水、无氧、碱性条件下与溴乙烷反应生成4-溴苯乙醚brph,反应时间为12小时;
[0012]
步骤(ii)为制备芴单元修饰性官能团的步骤,具体为,在无水无氧条件下芴酮和
过量的所述4-溴苯乙醚的格式反应,反应温度为65℃,反应时间为12小时,生成中间产物即芴单元修饰性官能团fphoh;
[0013]
步骤(iii)为制备氰基修饰的联咔唑中心的前驱体的步骤,具体为,在碱性条件下的三溴咔唑和2-氟苯腈进行反应,反应温度为150℃,反应时间为12小时,生成中间产物即氰基修饰的联咔唑中心的前驱体brczphcn;
[0014]
步骤(iv)具体为,在催化剂1,1'-双二苯基膦二茂铁二氯化钯条件下通过brczphcn与联硼酸频那醇酯在110℃下反应12小时生成中间产物bczphcn,;
[0015]
步骤(v)具体为,通过所述brczphcn和所述bczphcn的suzuki偶联反应生成具有联咔唑的中间产物dczphcn,催化剂使用的是四(三苯基膦)钯,在110℃下反应12小时;
[0016]
步骤(vi)为酸催化的friedel-crafts反应,具体为,所述dczphcn和所述fphoh在室温条件下合成本发明所述产物dfczphcn,反应时间为3小时。
[0017]
本发明合成的具有联咔唑双极中心的化合物dfczphcn,在氰基、芴单元以及烷基链的修饰下有效调节了化合物的共轭结构以及电子云的分布状态,使得整个结构在被光激发后产生分子内电荷转移(ict);化合物具有强烈的紫外吸收并且在422nm出表现出强烈的蓝色荧光发射,同时其重量损失5%时温度可以达到462℃,具有优异的热稳定性能,可用于oled器件发光层材料或者作为蓝光的主体材料的器件;
[0018]
本发明合成的化合物dfczphcn在联咔唑的基础上引入芴单元以及拟卤素氰基基团,氰基对联咔唑类双极性材料的发光和电荷传输性能带来了明显的影响,双极中心既可以进行电荷传输,也可以进行空穴传输,因此还可以作为电子或空穴载流子传输材料。
[0019]
有益效果:
[0020]
1.合成原料价格便宜,易于获得,其成本低廉。
[0021]
2.由于芴单元的存在,有效调控了材料荧光发射光谱的红移,实现了强的紫外-可见吸收以及在422nm强烈的蓝色荧光发射,适用于作为蓝光oled器件的发光层材料。
[0022]
3.由于氰基的碳氮三键的存在以及咔唑3号位和6号位上的共轭延伸,材料重量损失5%时温度可以达到462℃,具有非常优异的热稳定性能。
附图说明
[0023]
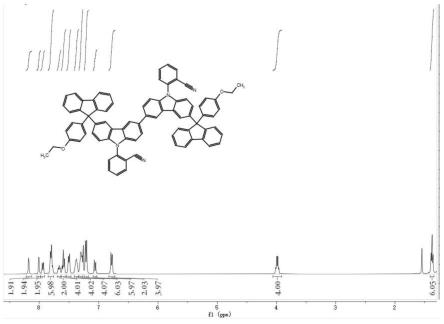
图1实施例1制备的dfczphcn的1hnmr谱图;
[0024]
图2实施例1制备的dfczphcn的飞行时间质谱图;
[0025]
图3实施例1制备的dfczphcn的dsc测试曲线;
[0026]
图4实施例1制备的dfczphcn的tga测试曲线;
[0027]
图5实施例1制备的dfczphcn在甲苯溶液中的紫外-可见吸收光谱;
[0028]
图6实施例1制备的dfczphcn和比较例1制备的dczphcn在甲苯溶液中的荧光发射光谱;
[0029]
图7实施例1制备的dfczphcn在二氯甲烷溶液中的电化学氧化曲线。
具体实施方案
[0030]
为了更好理解本发明专利的具体内容,下面将通过详细的实验步骤来进一步说明本发明的合成技术方案与路线。具体包括合成路线以及相关性质表征。但是实施例并不限
制本发明。
[0031]
实施例1:化合物dfczphcn的合成
[0032]
化合物dfczphcn的结构如图所示:
[0033][0034]
化合物dfczphcn的制备过程如下:
[0035]
步骤(i),brph的合成
[0036]
向100ml反应瓶中加入对溴苯酚(4.35g,25.00mmol)、碳酸钾(4.14g,50.00mmol),橡胶塞密封,抽真空和补充氮气重复三次,向反应瓶中加入氮气球鼓泡后的n,n-二甲基甲酰胺溶液(30m)使其充分溶解,再在氮气环境下加入溴乙烷(3.27g,30.00mmol),将油浴锅温度升高至80℃,冷凝回流8小时。使用tcl薄层色谱检测,二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除去溶剂,使用石油醚:二氯甲烷(体积比为15:1)洗脱剂,在硅胶柱中进行柱层析法分离提纯,后得淡黄色透明油状液体3.41g(产率:68%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ7.39
–
7.34(m,2h),6.79
–
6.74(m,2h),3.99(q,j=7.0hz,2h),1.41(td,j=6.9,0.5hz,3h).
[0037]
步骤(ii),fphoh的合成
[0038]
向装有磁子的50ml反应瓶中加入打磨过的镁屑(0.192g,8mmol),一粒碘,橡胶塞密封,抽真空和补充氮气重复三次,取化合物brph(1.00g,5mmol)和除水处理过的四氢呋喃(5ml)溶剂缓慢加入反应瓶之中,同时用热吹风机对其加热,直至棕黄色的溶液转至透明无色溶液说明引发成功,停止加热。将反应装置转移至预热好的油浴锅中,再将9-芴酮(0.90g,5mmol)溶解于四氢呋喃(5ml)溶液中,于氮气环境下加入反应瓶,于65℃下冷凝回流12h,使用tcl薄层色谱检测,加入饱和氯化铵水溶液(6ml)猝灭反应。饱和食盐水洗涤并用二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除去溶剂,使用石油醚:二氯甲烷(体积比为3:1)洗脱剂,在硅胶柱中进行柱层析法分离提纯,后得淡黄色固体粉末0.97g(产率:64.3%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ7.68(d,j=7.5hz,2h),7.48
–
7.19(m,8h),6.90
–
6.74(m,2h),4.00(q,j=7.0hz,2h),2.68(s,1h),1.41(t,j=7.0hz,3h).
[0039]
步骤(iii),brczphcn的合成
[0040]
向100ml反应瓶中加入3-溴咔唑(0.74g,3.00mmol)、碳酸铯(1.40g,4.00mmol),使用橡胶塞密封,抽真空并补充氮气循环三次,向反应瓶中加入氮气球鼓泡后的n,n-二甲基甲酰胺溶液(30ml)使其充分溶解,再在氮气环境下加入2-氟苯腈(0.49g,4.00mmol),将油浴锅温度升高至160℃,冷凝回流12小时。使用tcl薄层色谱检测,待反应降至室温后使用饱和食盐水洗涤并用二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除
去溶剂,使用石油醚:二氯甲烷(体积比为5:1)洗脱剂,在硅胶柱中进行柱层析法分离提纯,分离后获得白色片状固体0.63g(产率:78%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ8.27(d,j=1.9hz,1h),8.10(d,j=7.8hz,1h),7.94(dd,j=7.8,1.6hz,1h),7.82(td,j=7.8,1.6hz,1h),7.66
–
7.56(m,2h),7.55
–
7.44(m,2h),7.35(t,j=7.5hz,1h),7.20(d,j=8.2hz,1h),7.08(d,j=8.7hz,1h).
[0041]
步骤(iv),bczphcn的合成
[0042]
向50ml两口反应瓶中加入brczphcn(0.52g,1.50mmol)、联硼酸频哪醇酯(0.57g,2.25mmol)、乙酸钾(0.45g,4.50mmol)、使用橡胶塞密封,抽真空和补充氮气重复三次,在氮气环境下加入pd(dppf)cl2(0.0525g,0.075mmol),再向反应瓶中加入鼓氮气处理后的1,4-二氧六环溶液(25ml)使其充分溶解,将油浴锅温度升高至110℃,冷凝回流12小时。使用tcl薄层色谱检测,待反应降至室温后使用饱和食盐水洗涤并用二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除去溶剂,使用石油醚:二氯甲烷(体积比为3:1)洗脱剂,在硅胶柱中进行柱层析法分离提纯,后得白色固体粉末0.41g(产率:67%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ8.65(s,2h),8.19(d,j=7.7hz,2h),7.95(d,j=7.7hz,2h),7.92
–
7.80(m,4h),7.63(t,j=8.1hz,4h),7.43(t,j=7.7hz,2h),7.34(t,j=7.4hz,2h),7.26(s,1h),7.19(dd,j=8.3,3.0hz,4h).
[0043]
步骤(v),dczphcn的合成
[0044]
向50ml反应瓶中加入brczphcn(0.29g,0.84mmol)、化合物bczphcn(0.40g,0.10mmol)、使用橡胶塞密封,抽真空和补充氮气重复三次,在氮气环境下加入四三苯基膦钯(0.097g,0.084mmol),再向反应瓶中加入鼓氮气处理后的2mol/l碳酸钾(5ml)、甲苯(10ml)、乙醇(5ml)的混合溶液使其充分溶解,将油浴锅温度升高至110℃,冷凝回流12小时。使用tcl薄层色谱检测,待反应降至室温后使用饱和食盐水洗涤并用二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除去溶剂,使用石油醚:二氯甲烷(体积比为2:1)洗脱剂,在硅胶柱中进行柱层析法分离提纯,后得白色固体粉末0.22g(产率:51.3%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ8.40
–
8.32(m,2h),8.15(dd,j=7.8,3.2hz,2h),7.88(d,j=7.8hz,2h),7.75(t,j=7.8hz,2h),7.68(dd,j=7.9,5.6,1.8hz,2h),7.60
–
7.50(m,4h),7.37(t,j=7.7hz,2h),7.28(d,j=7.5hz,2h),7.22(d,j=8.5hz,2h),7.15(d,j=8.1hz,2h).maldl-tof-ms:m/z calcd for c
38h22
n4[m]
+
:534.62,found:533.987.
[0045]
步骤(vi),dfczphcn的合成
[0046]
向装有磁子的50ml反应瓶中加入dczphcn(0.20g,0.40mmol),加入二氯甲烷(20ml)溶解,再加入三氟化硼乙醚(0.128g,0.80mmol),再将化合物fphoh(0.302g,1.00mmol)溶于二氯甲烷溶液(20ml)以每秒2滴的速度缓慢加入反应瓶,在常温下搅拌3小时。使用tcl薄层色谱检测,加水猝灭多余的酸,使用饱和食盐水洗涤并用二氯甲烷萃取三次,将收集的下层有机相用无水na2so4干燥,减压蒸馏除去溶剂,使用石油醚:二氯甲烷(体积比为2:3)洗脱剂,在硅胶柱中进行柱层析法分离提纯,后得白色固体粉末0.24g(产率:54.0%)。产物核磁:1h nmr(400mhz,cdcl3,ppm)δ8.18(s,2h),8.01(s,2h),7.94(d,j=7.7hz,2h),7.80(d,j=7.6hz,6h),7.67(t,j=7.2hz,2h),7.60(t,j=8.0hz,4h),7.50(d,j=7.6hz,4h),7.38(p,j=6.1hz,4h),7.30(s,6h),7.21(d,j=8.4hz,6h),7.06(d,j=
8.7hz,2h),6.78(d,j=8.3hz,4h),3.99(q,j=6.9hz,4h),1.38(t,j=6.9hz,6h).maldl-tof-ms:m/z calcd for c
80h54
n4o2[m]
+
:1103.34,found:1102.70.
[0047]
比较例1:dczphcn的合成
[0048]
比较例1中间体dczphcn的结构如图所示:
[0049][0050]
比较例1中间体dczphcn的合成步骤如实施例1中步骤(i)-步骤(v),操作完全相同。
[0051]
物理性能测试
[0052]
通过400mhz1h nmr核磁共振波谱仪和maldi-tof-ms飞行时间质谱对产物的结构和相对分子质量进行了表征。热稳定分析通过dsc差示扫描量热仪和同步热分析仪在氮气氛围下进行测量。通过紫外-可见分光光度计lambda-35、荧光分光光度计rf-6000分析测定了dfczphcn的光谱性质。通过chi660e电化学工作站的循环伏安法测试了化合物的电化学性质。
[0053]
称取5mg dfczphcn溶解于0.6ml氘带氯仿中,通过核磁共振波谱仪进行分析测试,得到dfczphcn的1hnmr谱图,测试结果如图1所示。
[0054]
称取1mg dfczphcn溶解于二氯甲烷中,通过飞行时间质谱仪maldi-tof-ms进行分析测试,得到dfczphcn的maldi-tof谱图,测试结果如图2所示。
[0055]
称取5mg dfczphcn,通过dsc差示扫描量热仪在氮气下进行测试,得到dfczphcn的dsc测试曲线,测试结果如图3所示。dfczphcn的玻璃化转变温度为141℃,表明其具有较好的成膜性能。其熔点可达到225℃,表明材料具有良好的热稳定性。
[0056]
称取5mg dfczphcn,通过同步热分析仪在氮气氛围下进行测试,得到dfczphcn的tga测试曲线,测试结果如图4所示。dfczphcn的5%质量分解温度td为462℃,这得益于二联咔唑以及芴单元之间的刚性共轭结构,使得dfczphcn具有非常高的热分解温度,这有利于提高有机发光二极管器件的寿命以及稳定性。
[0057]
称取5.5mg实施1制备的dfczphcn溶解于甲苯中,配制成浓度为1
×
10-5
mol/l的溶液,通过lambda-35型号紫外-可见分光光度计进行分析测试,得到dfczphcn的紫外-可见吸收光谱,测试结果如图5所示。在溶液状态下,化合物在300nm处的吸收是由于联咔唑骨架部分的n-π*跃迁导致的,320nm后较长的肩缝吸收可以归因于联咔唑主体到缺电子氰基苯的分子内电荷转移。通过光谱推导出其光学带隙eg为3.36ev。
[0058]
称取5.5mg实施1制备的dfczphcn溶解于甲苯中,配制成浓度为1
×
10-5
mol/l的溶液,通过rf-6000型号荧光分光光度计进行分析测试,得到dfczphcn的荧光发射光谱,测试
结果如图6所示。称取2.7mg比较例1制备的dczphcn溶解于甲苯中,配制成浓度为1
×
10-5
mol/l的溶液,通过荧光分光光度计rf-6000进行分析测试,得到dczphcn的荧光发射光谱,测试结果如图6所示,其结果同实施例1制备的dfczphcn的发射光谱结果相比可以看出,引入芴基单元后的dfczphcn最大荧光发射波长红移了11nm,实现了发射光谱的可调控性,达到了422nm的深蓝光发射,因此,dfczphcn可作为深蓝色磷光oled的发光层主体材料。
[0059]
称取5mg实施1制备的dfczphcn溶解于无水无氧处理的二氯甲烷中,电解质为四丁基六氟磷酸铵,通过chi660e电化学工作进行分析测试,得到dfczphcn的电化学氧化曲线,测试结果如图7所示。表现出可逆的氧化过程,说明具有良好的电化学稳定性。结合光谱数据dfczphcn可知其homo能级为-5.79ev,其lumo能级为-2.43ev,在数值上来说,其能级可以和阳极ito层以及常见的电子输入层的材料的功函数值达到较高的匹配度,有利于其实现电子传输过程;同时联咔唑中心又具备空穴传输性能,dfczphcn可同时具备电荷和空穴的双重传输性能,有望成为双极性传输材料。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1