化合物Tomivosertib的制备方法与流程
化合物tomivosertib的制备方法
技术领域
1.本发明涉及生物医药技术领域,具体地,涉及一种激酶抑制剂化合物tomivosertib的制备方法。
背景技术:
2.effector治疗公司正在开发tomivosertib(eft-508),一种有效的mnk1和mnk2激酶选择性抑制剂,用于癌症的口服治疗。目前,其临床试验适应症覆盖了:弥漫性大b细胞淋巴瘤、头颈部肿瘤、肝细胞癌、激素难治性前列腺癌、淋巴瘤、转移性乳腺癌、转移性结直肠癌、非小细胞肺癌、移行细胞癌、实体瘤,这些适应症目前均处于2期临床研究阶段,其中除转移性乳腺癌在加拿大处于2期临床研究外,其余适应症均在美国处于2期临床研究。
3.tomivosertib的化学结构具体如式i所示化合物:
[0004][0005]
然而,目前化合物tomivosertib的新的制备方法仍有待改进。
技术实现要素:
[0006]
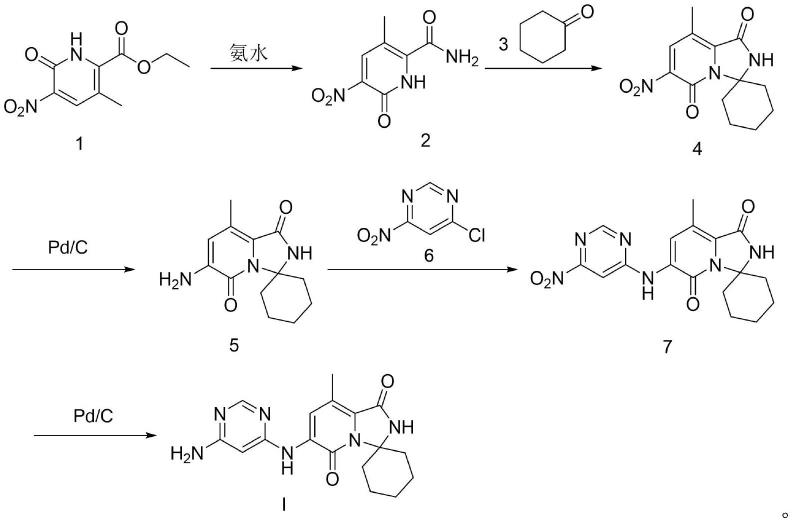
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出一种化合物tomivosertib新的制备方法。相比于现有技术,本发明的制备方法,采用商售原料,经5步反应制备得到,其路线总体产率较高,反应条件温和,操作简易,纯化过程简单,适合于原料药的工业化大生产的需求。其中步骤(1)利用氨水脱除羧酸保护基后同时进行酰胺化反应,减少了反应步骤,同时具有较高的产率,且采用封管作为反应容器,可以有效缩短反应时间,提高反应效率。在步骤(2)中加入环己酮(式3所示化合物),然后分子内脱水缩合成环得到目标产物,相比于其他文献采用卤代反应生成产物时选择性较低的问题,该方法不仅简化了反应步骤,还能提高反应产率,有利于反应纯化。步骤(4)通过加入催化剂和配体进行特异性的卤代反应,有效提高了反应的选择性,减少了副反应。
[0007]
在本发明的一个方面,本发明提供了一种式i所示化合物tomivosertib的制备方法。根据本发明的实施例,该方法包括:
[0008]
(1)使式1所示化合物与氨水接触,以便获得式2所示化合物;
[0009]
(2)使式2所示化合物与环己酮(式3所示化合物)接触,以便获得式4所示化合物;
[0010]
(3)使式4所示化合物与pd/c接触,以便获得式5所示化合物;
[0011]
(4)使式5所示化合物与式6所示化合物接触,以便获得式7所示化合物;
[0012]
(5)使式7所示化合物与pd/c接触,以便获得式i所示化合物,
[0013][0014]
本发明所述的合成路线采用商售原料(其中式1所示化合物的cas号为1509897-31-7,式3所示化合物的cas号为108-94-1,式6所示化合物的cas号为126827-19-8),经5步反应制备得到式i所示化合物tomivosertib。发明人发现,本发明的制备方法,其路线总体产率较高,反应条件温和,操作简易,纯化过程简单,适合于原料药的工业化大生产的需求。
[0015]
在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至少两种反应物发生化学反应的方式,例如可以是将两种反应物在适当的条件下进行混合。根据需要,可以在搅拌下,将需要进行接触的反应物进行混合,由此,搅拌的类型并不受特别限制,例如可以为机械搅拌,即在机械力的作用下进行搅拌。
[0016]
在本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
[0017]
根据本发明的实施例,上述制备式2所示化合物、式4所示化合物、式5所示化合物、式7所示化合物、式i所示化合物的方法还可以具有下列附加技术特征至少之一:
[0018]
根据本发明的实施例,本发明所述的化学反应可以按照本领域已知的任何方法进行。制备式2所示化合物、式4所示化合物、式5所示化合物、式7所示化合物、式i所示化合物的原料的来源并不受特别限制,其可以是采用任何已知的方法制备的,或者市售获得的。
[0019]
根据本发明的实施例,在步骤(1)中,使式1所示化合物与氨水的接触方式并不受特别限制。由此,可以提升式1所示化合物与氨水接触反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
[0020]
根据本发明的实施例,在步骤(1)中,包括如下步骤:室温下,将式1所示化合物加入甲醇中,再加入氨水于密封管中,将混合物搅拌并在60℃~68℃加热反应2小时45分钟~3小时30分钟,tlc显示原料完全消耗,将反应液冷却至室温,混合液用乙酸乙酯萃取三次,合并有机相后用盐水洗涤后浓缩,浓缩物用体积比为(8~12):1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式2所示化合物。由此,可以提升式1所示化合物和氨水接触
反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
[0021]
根据本发明的实施例,在步骤(1)中,式1所示化合物和氨水的质量体积比为1:(30~40)(g/v),优选式1所示化合物和氨水的质量体积比为1:36。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0022]
根据本发明的实施例,在步骤(1)中,优选式1所示化合物和氨水反应的温度为65℃,反应时间为3小时。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0023]
根据本发明的实施例,在步骤(1)中,优选所述硅胶柱层析的溶剂为体积比为10:1的石油醚/乙酸乙酯混合溶剂。
[0024]
根据本发明的一个具体实施例,在步骤(1)中,包括如下步骤:室温下,将式1所示化合物(5.0g,0.0221mol)加入甲醇(15ml)中,再加入氨水(180ml)于500ml的密封管中,将混合物搅拌并在65℃加热反应3小时,tlc显示原料完全消耗,将反应液冷却至室温,混合液用乙酸乙酯(3
×
90ml)萃取三次,合并有机相后用盐水(90ml)洗涤后浓缩,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式2所示化合物,得量4.0g,收率91.8%。
[0025]
根据本发明的实施例,在步骤(2)中,式2所示化合物与式3所示化合物、以及盐酸二氧六环溶液的接触方式并不受特别限制。由此,可以提升式2所示化合物与式3所示化合物、以及盐酸二氧六环溶液接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
[0026]
根据本发明的实施例,在步骤(2)中,包括如下步骤:室温下在封管中,向式2所示化合物和式3所示化合物的无水二氧六环溶液中,再加入4m盐酸二氧六环溶液,然后加热升温至100℃,封管中反应10小时,tlc显示原料完全消耗,反应溶液减压浓缩后,浓缩物用体积比为(8~12):1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式4所示化合物。由此,可以提升式2所示化合物与式3所示化合物、以及盐酸二氧六环溶液接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
[0027]
根据本发明的实施例,在步骤(2)中,4m盐酸二氧六环溶液的制取方法为:称取定量的二氧六环,通干燥hcl气体,称重至计算量,停止通气即可得到。
[0028]
根据本发明的实施例,在步骤(2)中,式2所示化合物与式3所示化合物反应的摩尔比为1:(1.0~1.2),优选式2所示化合物与式3所示化合物反应的摩尔比为1:1.0。由此,可以进一步提高利用该方法制备式4所示化合物的效率。
[0029]
根据本发明的实施例,在步骤(2)中,式2所示化合物与无水二氧六环溶液、以及盐酸二氧六环溶液的质量体积比为1:10:10。由此,可以进一步提高利用该方法制备式4所示化合物的效率。
[0030]
根据本发明的实施例,在步骤(2)中,优选所述硅胶柱层析的溶剂为体积比为10:1的石油醚/乙酸乙酯混合溶剂。
[0031]
根据本发明的一个具体实施例,在步骤(2)中,包括如下步骤:室温下在封管中,向式2所示化合物(19.7g,0.1mol)和式3所示化合物(9.8g,0.1mol)的无水二氧六环溶液(200ml)中,再加入4m盐酸二氧六环溶液(200ml),然后加热升温至100℃,封管中反应10小时,tlc显示原料完全消耗,反应溶液减压浓缩后,浓缩物用体积比为10:1的石油醚/乙酸乙
酯混合溶剂在硅胶上柱层析纯化产物,得式4所示化合物,得量23.7g,收率85.5%。
[0032]
根据本发明的实施例,在步骤(3)中,式4所示化合物与pd/c的接触方式并不受特别限制。由此,可以提升式4所示化合物与pd/c接触反应的效率,加快反应速度,进一步提高利用该方法制备式5所示化合物的效率。
[0033]
根据本发明的实施例,在步骤(3)中,包括如下步骤:氮气保护下向含式4所示化合物的甲醇溶液中加入pd/c,用氢气在真空下置换2~5次,反应液保持在2.0~3.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式5所示化合物的粗品固体。由此,可以提升式4所示化合物与pd/c接触反应的效率,加快反应速度,进一步提高利用该方法制备式5所示化合物的效率。
[0034]
根据本发明的实施例,在步骤(3)中,所述pd/c为5%~20%pd/c,优选所述pd/c为5%pd/c、10%pd/c、或20%pd/c。5%~20%pd/c是作为催化氢化反应的催化剂,例如5%pd/c指在此pd/c混合物中金属pd的含量为5%,10%pd/c指在此pd/c混合物中金属pd的含量为10%。
[0035]
根据本发明的实施例,在步骤(3)中,优选式4所示化合物与所述10%pd/c反应的质量比为1:0.1。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
[0036]
根据本发明的实施例,在步骤(3)中,优选式4所示化合物与所述5%pd/c反应的质量比为1:0.2。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
[0037]
根据本发明的实施例,在步骤(3)中,优选式4所示化合物与所述20%pd/c反应的质量比为1:0.05。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
[0038]
根据本发明的一个具体实施例,在步骤(3)中,包括如下步骤:氮气保护下向含式4所示化合物(3.8g,0.0137mol)的甲醇(150ml)溶液中加入10%pd/c(380mg),用氢气在真空下置换3次,反应液保持在2.5个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式5所示化合物的粗品固体3.25g,收率95.9%,粗品直接可用于下一步反应。
[0039]
根据本发明的实施例,在步骤(4)中,式5所示化合物与式6所示化合物、以及与pd(oac)2、xantphos、cs2co3的接触方式并不受特别限制。由此,可以提升式5所示化合物与式6所示化合物、以及与pd(oac)2、xantphos、cs2co3接触反应的效率,加快反应速度,进一步提高利用该方法制备式7所示化合物的效率。
[0040]
根据本发明的实施例,在步骤(4)中,包括如下步骤:室温下,将式5所示化合物和式6所示化合物加入1,4-二氧六环中,搅拌均匀,加入pd(oac)2、xantphos和cs2co3,将反应液在氮气保护下,升温保持在96℃~100℃反应4h,反应毕,将反应液过滤后减压浓缩,浓缩物用体积比为(8~12):1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式7所示化合物。由此,可以提升式5所示化合物与式6所示化合物、以及与pd(oac)2、xantphos、cs2co3接触反应的效率,加快反应速度,进一步提高利用该方法制备式7所示化合物的效率。
[0041]
根据本发明的实施例,在步骤(4)中,式5所示化合物与式6所示化合物、以及与pd(oac)2、xantphos、cs2co3反应的摩尔比为1:(1.0~1.2):(0.12~0.2):(0.12~0.2):(2.5~5),优选式5所示化合物与式6所示化合物、以及与pd(oac)2、xantphos、cs2co3反应的摩尔比为1:1.05:0.15:0.15:3.6。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0042]
根据本发明的实施例,在步骤(4)中,优选所述硅胶柱层析的溶剂为体积比为10:1的石油醚/乙酸乙酯混合溶剂。
[0043]
根据本发明的一个具体实施例,在步骤(4)中,包括如下步骤:室温下,将式5所示化合物(24.7g,0.1mol)和式6所示化合物(16.7g,0.105mol)加入1,4-二氧六环(500ml)中,搅拌均匀,加入pd(oac)2(3.4g,0.015mol),xantphos(14.4g,0.015mol)和cs2co3(117.3g,0.36mol),将反应液在氮气保护下,升温保持在100℃反应4h,反应毕,将反应液过滤后减压浓缩,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式7所示化合物,得量34.6g,收率93.4%。
[0044]
根据本发明的实施例,在步骤(5)中,式7所示化合物与pd/c的接触方式并不受特别限制。由此,可以提升式7所示化合物与pd/c接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
[0045]
根据本发明的实施例,在步骤(5)中,包括如下步骤:氮气保护下向含式7所示化合物的甲醇溶液中加入pd/c,用氢气在真空下置换2~5次,反应液保持在2.0~3.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式i所示化合物的粗品固体,粗品经再重结晶得到式i所示化合物的精制品固体。由此,可以提升式7所示化合物与pd/c接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
[0046]
根据本发明的实施例,在步骤(5)中,所述pd/c为5%~20%pd/c,优选所述pd/c为5%pd/c、10%pd/c、或20%pd/c。在本发明中,5%~20%pd/c是作为催化氢化反应的催化剂,例如5%pd/c指在此pd/c混合物中金属pd的含量为5%,10%pd/c指在此pd/c混合物中金属pd的含量为10%。
[0047]
根据本发明的实施例,在步骤(5)中,优选式7所示化合物与所述10%pd/c反应的质量比为1:0.1。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
[0048]
根据本发明的实施例,在步骤(5)中,优选式7所示化合物与所述5%pd/c反应的质量比为1:0.2。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
[0049]
根据本发明的实施例,在步骤(5)中,优选式7所示化合物与所述20%pd/c反应的质量比为1:0.05。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
[0050]
根据本发明的一个具体实施例,在步骤(5)中,包括如下步骤:氮气保护下向含式7所示化合物(3.0g,0.0081mol)的甲醇(30ml)溶液中加入10%pd/c(300mg),用氢气在真空下置换3次,反应液保持在2.5个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式i所示化合物的粗品固体,粗品经再重结晶(粗品重结晶的溶剂是石油醚/乙酸乙酯=5/1,粗品约2.8g,溶剂的用量比例是1g/10ml(用的结晶溶剂28ml))得到式i所示化合物的精制品固体,得量2.50g,收率90.7%,hplc纯度99.8%。
[0051]
根据本发明的实施例,式i所示化合物的合成路线可以如下所示:
[0052][0053]
相对于现有技术,本发明所述的制备方法,其至少具有以下有益效果:本发明所述的方法,利用商售原料经过5步反应步骤制备得到式i所示化合物tomivosertib。其中步骤1利用氨水脱除羧酸保护基后同时进行酰胺化反应,减少了反应步骤,同时具有较高的产率。采用封管作为反应容器,可以有效缩短反应时间,提高反应效率。步骤2中加入环己酮进行羰基亲核加成反应,然后分子内脱水缩合成环得到目标产物,相比于其他文献采用卤代反应生成产物时选择性较低的问题,该方法不仅简化了反应步骤,还能提高反应产率,有利于反应纯化。步骤3是硝基还原反应。步骤4通过加入催化剂和配体进行特异性的卤代反应,有效提高了反应的选择性,减少了副反应。步骤3是硝基还原反应,无需进行纯化。本发明所述的制备方法,5步反应中有2步(步骤3和步骤5)属于硝基还原反应,原料选择硝基化合物可以减少反应中的位点,选择性的进行环化和卤代反应,可以有效提高每步的反应产率;路线总体产率较高,反应条件温和,操作简易,纯化过程简单,适合于原料药的工业化大生产的需求。
具体实施方式
[0054]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0055]
实施例1式2所示化合物的合成
[0056]
室温下,将式1所示化合物(5.0g,0.0221mol)加入甲醇(15ml)中,再加入氨水(180ml)于500ml的密封管中,将混合物搅拌并在65℃加热反应3小时,tlc显示原料完全消耗,将反应液冷却至室温,混合液用乙酸乙酯(3
×
90ml)萃取三次,合并有机相后用盐水(90ml)洗涤后浓缩,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式2所示化合物,得量4.0g,收率91.8%。
[0057]
lc-ms(apci):m/z=198.2(m+1)
+
。
[0058]
实施例2式2所示化合物的合成
[0059]
室温下,将式1所示化合物(5.0g,0.0221mol)加入甲醇(15ml)中,再加入氨水(150ml)于500ml的密封管中,将混合物搅拌并在60℃加热反应3小时30分钟,tlc显示原料完全消耗,将反应液冷却至室温,混合液用乙酸乙酯(3
×
90ml)萃取三次,合并有机相后用盐水(90ml)洗涤后浓缩,浓缩物用体积比为8:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式2所示化合物,得量3.8g,收率87.2%。
[0060]
实施例3式2所示化合物的合成
[0061]
室温下,将式1所示化合物(5.0g,0.0221mol)加入甲醇(15ml)中,再加入氨水(200ml)于500ml的密封管中,将混合物搅拌并在68℃加热反应2小时45分钟,tlc显示原料完全消耗,将反应液冷却至室温,混合液用乙酸乙酯(3
×
90ml)萃取三次,合并有机相后用盐水(90ml)洗涤后浓缩,浓缩物用体积比为12:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式2所示化合物,得量3.9g,收率89.5%。
[0062]
实施例4式4所示化合物的合成
[0063]
室温下在封管中,向式2所示化合物(19.7g,0.1mol)和式3所示化合物(9.8g,0.1mol)的无水二氧六环溶液(200ml)中,再加入4m盐酸二氧六环溶液(200ml),然后加热升温至100℃,封管中反应10小时,tlc显示原料完全消耗,反应溶液减压浓缩后,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式4所示化合物,得量23.7g,收率85.5%。
[0064]
lc-ms(apci):m/z=278.3(m+1)
+
。
[0065]
实施例5式4所示化合物的合成
[0066]
室温下在封管中,向式2所示化合物(19.7g,0.1mol)和式3所示化合物(10.3g,0.105mol)的无水二氧六环溶液(200ml)中,再加入4m盐酸二氧六环溶液(200ml),然后加热升温至100℃,封管中反应10小时,tlc显示原料完全消耗,反应溶液减压浓缩后,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式4所示化合物,得量23.6g,收率85.1%。
[0067]
实施例6式4所示化合物的合成
[0068]
室温下在封管中,向式2所示化合物(19.7g,0.1mol)和式3所示化合物(11.8g,0.12mol)的无水二氧六环溶液(200ml)中,再加入4m盐酸二氧六环溶液(200ml),然后加热升温至100℃,封管中反应10小时,tlc显示原料完全消耗,反应溶液减压浓缩后,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式4所示化合物,得量22.7g,收率81.9%。
[0069]
实施例7式5所示化合物的合成
[0070]
氮气保护下向含式4所示化合物(3.8g,0.0137mol)的甲醇(150ml)溶液中加入10%pd/c(380mg),用氢气在真空下置换3次,反应液保持在2.5个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式5所示化合物的粗品固体3.25g,收率95.9%,粗品直接可用于下一步反应。
[0071]
lc-ms(apci):m/z=248.3(m+1)
+
。
[0072]
实施例8式5所示化合物的合成
[0073]
氮气保护下向含式4所示化合物(3.8g,0.0137mol)的甲醇(150ml)溶液中加入5%
pd/c(760mg),用氢气在真空下置换2次,反应液保持在2.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式5所示化合物的粗品固体3.21g,收率94.8%,粗品直接可用于下一步反应。
[0074]
实施例9式5所示化合物的合成
[0075]
氮气保护下向含式4所示化合物(3.8g,0.0137mol)的甲醇(150ml)溶液中加入20%pd/c(190mg),用氢气在真空下置换5次,反应液保持在3.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式5所示化合物的粗品固体3.22g,收率95.1%,粗品直接可用于下一步反应。
[0076]
实施例10式7所示化合物的合成
[0077]
室温下,将式5所示化合物(24.7g,0.1mol)和式6所示化合物(16.7g,0.105mol)加入1,4-二氧六环(500ml)中,搅拌均匀,加入pd(oac)2(3.4g,0.015mol),xantphos(14.4g,0.015mol)和cs2co3(117.3g,0.36mol),将反应液在氮气保护下,升温保持在100℃反应4h,反应毕,将反应液过滤后减压浓缩,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式7所示化合物,得量34.6g,收率93.4%。
[0078]
lc-ms(apci):m/z=371.3(m+1)
+
。
[0079]
实施例11式7所示化合物的合成
[0080]
室温下,将式5所示化合物(24.7g,0.1mol)和式6所示化合物(15.9g,0.10mol)加入1,4-二氧六环(500ml)中,搅拌均匀,加入pd(oac)2(2.7g,0.012mol),xantphos(11.5g,0.012mol)和cs2co3(81.5g,0.25mol),将反应液在氮气保护下,升温保持在96℃反应4h,反应毕,将反应液过滤后减压浓缩,浓缩物用体积比为8:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式7所示化合物,得量34.1g,收率92.1%。
[0081]
实施例12式7所示化合物的合成
[0082]
室温下,将式5所示化合物(24.7g,0.1mol)和式6所示化合物(19.1g,0.12mol)加入1,4-二氧六环(650ml)中,搅拌均匀,加入pd(oac)2(4.5g,0.02mol),xantphos(19.2g,0.02mol)和cs2co3(163.0g,0.5mol),将反应液在氮气保护下,升温保持在98℃反应4h,反应毕,将反应液过滤后减压浓缩,浓缩物用体积比为12:1的石油醚/乙酸乙酯混合溶剂在硅胶上柱层析纯化产物,得式7所示化合物,得量33.6g,收率90.7%。
[0083]
实施例13式i所示化合物的合成
[0084]
氮气保护下向含式7所示化合物(3.0g,0.0081mol)的甲醇(30ml)溶液中加入10%pd/c(300mg),用氢气在真空下置换3次,反应液保持在2.5个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式i所示化合物的粗品固体,粗品经再重结晶(粗品重结晶的溶剂是石油醚/乙酸乙酯=5/1(v/v),粗品约2.8g,溶剂的用量比例是1g/10ml(用的结晶溶剂28ml))得到式i所示化合物的精制品固体,得量2.53g,收率91.8%,hplc纯度99.8%。
[0085]
lc-ms(apci):m/z=341.2(m+1)
+
。
[0086]
实施例14式i所示化合物的合成
[0087]
氮气保护下向含式7所示化合物(3.0g,0.0081mol)的甲醇(30ml)溶液中加入20%pd/c(150mg),用氢气在真空下置换5次,反应液保持在3.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式i所示化合
物的粗品固体,粗品经再重结晶(重结晶方法同实施例13)得到式i所示化合物的精制品固体,得量2.50g,收率90.7%,hplc纯度99.5%。
[0088]
实施例15式i所示化合物的合成
[0089]
氮气保护下向含式7所示化合物(3.0g,0.0081mol)的甲醇(30ml)溶液中加入5%pd/c(600mg),用氢气在真空下置换2次,反应液保持在2.0个大气压下,保持20℃~25℃搅拌反应,tlc显示原料完全消耗后将反应液过滤除去pd/c,滤液浓缩至干,得到式i所示化合物的粗品固体,粗品经再重结晶(重结晶方法同实施例13)得到式i所示化合物的精制品固体,得量2.44g,收率88.7%,hplc纯度99.4%。
[0090]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0091]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1