一种亚磷酰胺单体以及纯化寡核苷酸的方法与流程
一种亚磷酰胺单体以及纯化寡核苷酸的方法
1.相关申请的交叉引用
2.本技术要求2021年4月19日提交的申请号为202110417554.1、发明名称 为“一种亚磷酰胺单体以及纯化寡核苷酸的方法”的中国专利申请的优先权, 其全部内容通过引入并入本文。
技术领域
3.本发明属于寡核苷酸合成领域,更具体地,本发明通常涉及一种亚磷酰 胺单体以及纯化寡核苷酸的方法。
背景技术:
4.现代的寡核苷酸合成主要采用可自动化的亚磷酰胺固相合成技术,每一 步的合成产率极限可以达到99.6%。尽管如此,随着寡核苷酸链长的增加, 杂质含量也会增加,例如以合成产率99%计算,对于20-mer碱基长度的寡核 苷酸链,通常产率在80%左右,杂质含量在20%左右,对于40-mer碱基长 度的寡核苷酸链,产率与杂质含量则分别为65%和35%左右。产生的杂质主 要来自合成及脱保护过程中产生的小分子、每一步偶联反应不完全造成的失 败序列、盖帽不完全造成的缺失序列以及多重偶联造成的插入序列等,其中, 失败序列是最主要的杂质。
5.为了除去杂质和纯化得到的寡核苷酸,目前有多种手段可以实现该目 的,常见的如体积排阻色谱法、聚丙烯酰胺凝胶电泳、高效液相色谱法(包 含反相hplc及离子交换hplc)、纯化柱层析等,还有新技术如基于亲和力 的纯化方法(包括生物素-亲和素亲和纯化以及氟原子亲和柱纯化)。然而, hplc需要特殊仪器,且产生大量有机废液(如纯化5mg的寡核苷酸,需要 500l的溶剂),且寡核苷酸的大规模纯化成本高昂;同时容易造成交叉污染, 如果需要避免交叉污染,则每一个不同序列都应配备单独的hplc制备柱, 从而大大增加了纯化的成本;聚丙烯酰胺凝胶电泳操作复杂,难以实现高通 量纯化;体积排阻色谱法和纯化柱层析可以实现小规模的高通量纯化,但也 难以实现大规模的寡核苷酸纯化;基于捕获和释放策略,理论上可以解决交 叉污染的问题,也可以实现大规模的寡核苷酸纯化,但目前报道的合成纯化 的总体回收率仅为17.5%。
6.因此,开发新型的适用这种策略的单体将大大提高寡核苷酸的纯化效 率,并降低纯化的成本。
技术实现要素:
7.本发明的目的在于克服现有技术方法在纯化寡核苷酸以及去除其中作为 最主要杂质的失败序列时存在成本高昂、难以实现高通量、或纯化回收率低 等缺陷,本发明提供一种能够大幅提高寡核苷酸的纯化效率和降低纯化成本 的方法,该方法使用含有能够进行聚合反应的甲基丙烯酰胺基团的亚磷酰胺 单体作为合成寡核苷酸的原料,具有合成纯化回收率高、设备及操作简单、 且无交叉污染等优点,本发明人也是基于上述发现完成了
本发明。
8.为了实现上述目的,在一方面,本发明提供了一种亚磷酰胺单体,所述 亚磷酰胺单体具有如下式i表示的结构,或其药学上可接受的盐,或其对映 异构体、非对映异构体、互变异构体或溶剂化物:
9.[式i]
[0010][0011]
其中,x为具有如下结构的刚性连接子:
[0012][0013]
r1和r2均为-o(ch2ch2o)nch3,且分别取代在中间苯环上的两个任意 不同位置;r3至r10各自独立地选自氢、羟基、氰基、卤素、或者取代或 未取代的氨基、c
1-c6烷基、c
1-c6烷氧基或c
3-c6环烷基;m为1至10的整 数;n为1至10的整数;并且y为亚磷酰胺修饰的核苷,且所述核苷上的氨 基任选地被酰化,其中所述核苷可以为核糖核苷或脱氧核糖核苷。在本发明 的一个优选实施方式中,所述核苷为核糖核苷,所述核糖核苷包括胞苷(c)、 尿苷(u)、腺苷(a)、胸苷(t)或鸟苷(g)。在本发明的一个优选实施方 式中,所述核苷为脱氧核糖核苷,所述脱氧核糖核苷包括脱氧胞苷(c)、脱 氧尿苷(u)、脱氧腺苷(a)、脱氧胸苷(t)或脱氧鸟苷(g)。
[0014]
如本文所用,术语“药学上可接受的盐”包括药学上可接受的酸加成盐 和药学上可接受的碱加成盐。其中,所述药学上可接受的酸加成盐是指能够 保留游离碱的生物有效性而无其它副作用的与无机酸或有机酸所形成的盐, 例如无机酸盐可以包括但不限于盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、磷酸 盐等;有机酸盐可以包括但不限于甲酸盐、乙酸盐、三氟乙酸盐、丙酸盐、 己酸盐、水杨酸盐等。药学上可接受的碱加成盐是指能够保持游离酸的生物 有效性而无其它副作用的与无机碱或有机碱所形成的盐,例如无机碱盐可以 包括但不限于钠盐、钾盐、锂盐、铵盐、钙盐、镁盐、铁盐、锌盐、铜盐、 锰盐、铝盐等;有机碱盐可以包括但不限于伯胺类、仲胺类及叔胺类盐。这 些盐可通过本专业已知的方法制备。
[0015]
如本文所用,术语“酰化”(或称为“酰基化”)是指在有机分子中的氮、 氧、碳或硫等原子上引入脂肪族酰基rco-或芳香族酰基arco-的反应,其 中本发明中适用的所述酰化可以包括但不限于甲酰化、乙酰化、丙酰化、异 丙酰化、苯酰化等;且常用的酰化剂可以包括例如酰氯、酸酐和羧酸,更具 体地,例如乙酰氯或乙酸酐等。
[0016]
关于本发明的刚性连接子的具体结构,在本发明的一个优选实施方式中, r1和r2可以为彼此邻位、间位或对位取代;r3至r10可以各自独立地选 自氢、羟基、氰基、卤素、或者取代或未取代的氨基、c1-c6烷基;m可以 为3至8(例如3、4、5、6、7或8)的整数;n可以为2至5(例如2、3、 4或5)的整数;并且y可以为亚磷酰胺修饰的脱氧核糖核苷,所述脱氧核 糖核苷可以包括脱氧胞苷(c)、脱氧尿苷(u)、脱氧腺苷(a)、脱氧胸苷(t)或脱氧鸟苷(g),且所
述核苷上的氨基可以任选地被酰化(并且在优 选的情况下,所述核苷上的氨基被酰化)。
[0017]
进一步地,在本发明的一个优选实施方式中,r1和r2可以为彼此对位 取代。在本发明的一个优选实施方式中,r3至r10可以各自独立地选自氢、 羟基、氰基、卤素、或者取代或未取代的氨基、甲基、乙基、丙基、异丙基。
[0018]
在本发明的一个优选实施方式中,所述取代可以为被卤素(例如氟、氯 或溴)、羟基、氨基、氰基、c
1-c3烷基(例如甲基、乙基、丙基或异丙基) 或c
1-c3卤代烷基(例如卤代甲基、卤代乙基或卤代丙基)取代,但不限于 此。另外,所述取代可以包括一个或多个取代,取代数量的上限可以取决于 氢的数量,即全部取代。进一步地,在本发明的一个优选实施方式中,r1 和r2为对位取代;r3至r10均选自氢;m为5;并且n为2。
[0019]
在本发明的一个优选实施方式中,所述亚磷酰胺修饰中的修饰基团具有 如下结构:
[0020][0021]
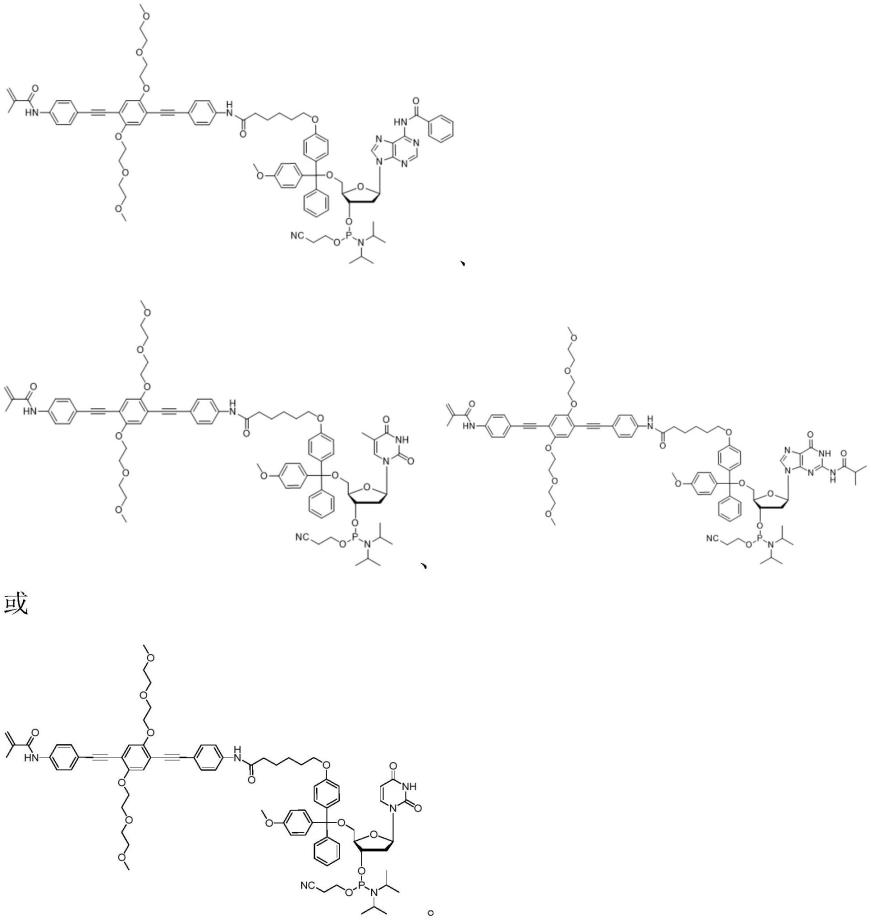
在本发明的一个优选实施方式中,所述亚磷酰胺单体可以进一步地表示 为具体的化合物,例如,其可以具有如下所示结构中的一种:
[0022]
[0023][0024]
在另一方面,本发明还提供了一种纯化寡核苷酸的方法,其特征在于, 所述方法包括以下步骤:(1)使寡核苷酸与上述亚磷酰胺单体发生偶联反应, 以使所述亚磷酰胺单体连接在所述寡核苷酸的末端,从而得到全长寡核苷酸; (2)聚合所述全长寡核苷酸;(3)从所聚合的全长寡核苷酸中去除失败序列; 以及(4)回收所述全长寡核苷酸。
[0025]
在根据本发明的纯化寡核苷酸的方法中,对于所述寡核苷酸的长度没有 特别的要求,可以根据实际需要进行选择。在本发明的一个实施方式中,所 述寡核苷酸的长度可以为10-100nt;优选为20-50nt(例如25nt、30nt、35nt 或40nt等)。
[0026]
对于步骤(1),即偶联步骤,所述偶联反应的时间可以为0.2h-1h(例如 0.3h、0.5h或0.8h等),优选为0.3h。在另一个实施方式中,所述偶联反应可 以分多次进行。例如,优选地,所述偶联可以分6次进行,每次0.05h(总计 0.3h)。经过偶联反应,所述亚磷酰胺单体可以连接在所述寡核苷酸的5’末端 或3’末端,但在优选的情况下,所述亚磷酰胺单体可以连接在所述寡核苷酸 的5’末端。
[0027]
对于步骤(2),所述聚合可以在存在交联剂的情况下进行,例如,在一 个实施方式
中,所述聚合可以通过使用包括n,n-二甲基丙烯酰胺和n,n'-亚 甲基双丙烯酰胺的试剂来进行。优选地,所述n,n-二甲基丙烯酰胺的浓度可 以为2-5m(例如2.5m、3m或4m等);更优选为3.7m;和/或所述n,n'-亚 甲基双丙烯酰胺的浓度可以为0.1-2m(例如0.5m、1m或1.5m等);更优选 为0.18m。另外,所述聚合还可以在也存在助聚剂的情况下进行,例如,在 一个实施方式中,所述聚合所使用的试剂还包括过硫酸铵和四甲基乙二胺。 优选地,所述过硫酸铵的质量体积浓度可以为1%-10%(例如3%、6%或8% 等);更优选为5%;和/或所述四甲基乙二胺的浓度可以为0.4m-1.0m(例如 0.5m、0.7m或0.9m等);优选为0.66m。
[0028]
对于步骤(3),所述去除失败序列可以通过依次执行切碎步骤(2)中聚 合寡核苷酸的凝胶、缓冲液浸泡和洗脱的步骤来进行。在一个优选的实施方 式中,所述缓冲液可以为纯水或3%-10%(例如4%、6%或8%等)的三乙胺 水溶液;更优选为5%的三乙胺水溶液。
[0029]
对于步骤(4),所述回收可以通过依次执行酸溶液浸泡、水溶液洗脱、 碱溶液中和以及浓缩的步骤来进行。在一个优选的实施方式中,所述酸溶液 可以为70%-95%(例如75%、80%、85%或95%等)的醋酸溶液;和/或所述 碱溶液可以为浓氨水(例如浓度大于15%的氨水溶液)。
[0030]
值得注意的是,本发明的方法使用含有能够进行聚合反应的甲基丙烯酰 胺基团的亚磷酰胺单体作为合成寡核苷酸的原料,该甲基丙烯酰胺结构在固 相合成、载体切割及碱基脱保护基团步骤中均可以稳定地连接于寡核苷酸链 上,并通过聚合反应使寡核苷酸链共价连接于固体高分子聚合物(如聚丙烯 酰胺凝胶)上。
[0031]
另外,根据本发明,在使用本发明所提供的亚磷酰胺单体进行寡核苷酸 纯化时,通常是在进行寡核苷酸链合成的最后一个循环中引入该亚磷酰胺单 体(优选地,引入在寡核苷酸链的5’端)。由于失败序列寡核苷酸链的5’位 羟基被固相合成中的盖帽步骤封闭而不能与本发明中的单体反应而连接于高 分子聚合物中,因而通过简单的洗涤高分子聚合物操作便可以纯化去除失败 序列,而全长的寡核苷酸链经简单的酸处理即可被释放,从而实现纯化的目 的。
[0032]
在另一方面,本发明还提供了一种试剂盒,其包含上述亚磷酰胺单体。
[0033]
在另一方面,本发明还提供了上述亚磷酰胺单体和上述试剂盒在寡核苷 酸纯化中的用途。
[0034]
基于本发明人的研究,本发明所提供的亚磷酰胺单体至少具有以下多个 优点:1)合成纯化回收率较高;2)需要的设备简单,只需要玻璃仪器;3) 原料便宜易得,聚合化学品为常规的聚丙烯酰胺凝胶所用化学品;4)操作简 单,只需要简单液体转移、过滤及洗涤操作;5)纯化时间短,不超过24小 时,轮转周期少;6)纯化规模不限,适合大规模寡核苷酸合成纯化;7)无 交叉污染,对于每一不同序列不同批次寡核苷酸都使用全新的共聚合物,因 此没有交叉污染;8)低能环保,纯化技术所需要的溶剂及缓冲液使用量小, 产生废液少,浓缩时间短,能耗低。
附图说明
[0035]
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与 下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在 附图中:
[0036]
图1示出了根据本发明的实施例1的方法制备亚磷酰胺单体的合成路线 图(其中,a)naoh,thf,2h;b)对二苯酚,k2co3,乙腈,72h;c)i2, kio3,acoh,48h;d)pd(pph3)4,cui,4-乙炔基苯胺,thf,et3n,24h; e)6-{4-[羟基-(4-甲氧基苯基)-苯基-甲基]苯氧基}己酸,diea,hatu,dcm,12h;f)diea,甲基丙烯酰氯,dcm,12h;g)乙酰氯,dc-ac,吡啶,16h; h)2-氰乙基n,n,n',n'-四异丙基亚磷酰二胺,btt,dcm,1h;以及i)乙酰 氯,dt,吡啶,16h);
[0037]
图2示出了粗品寡核苷酸的hplc图;
[0038]
图3示出了经单体t-8修饰后的寡核苷酸结构图;
[0039]
图4示出了200nmol合成规模纯化后寡核苷酸的hplc图(dc);
[0040]
图5示出了200nmol合成规模纯化后寡核苷酸纯品质谱图(dc);
[0041]
图6示出了1000nmol合成规模纯化后寡核苷酸的hplc图(dc);
[0042]
图7示出了1000nmol合成规模纯化后寡核苷酸纯品质谱图(dc);
[0043]
图8示出了200nmol合成规模纯化后寡核苷酸的hplc图(dt);
[0044]
图9示出了200nmol合成规模纯化后寡核苷酸纯品质谱图(dt);
[0045]
图10示出了1000nmol合成规模纯化后寡核苷酸的hplc图(dt);
[0046]
图11示出了1000nmol合成规模纯化后寡核苷酸纯品质谱图(dt);
[0047]
图12示出了捕获和释放策略纯化与hplc纯化引物用于pcr的产物胶 图(其中,m:标志物;1:t-8纯化的正向引物;2:hplc纯化的正向引物)
[0048]
图13示出了捕获和释放策略纯化与hplc纯化引物用于qpcr扩增的曲 线图;以及
[0049]
图14示出了捕获和释放策略纯化与hplc纯化引物用于qpcr的产物胶 图(其中,m:标志物;1:t-8纯化的正向引物;2:hplc纯化的正向引 物;3:t-8ntc;4:hplc ntc)。
具体实施方式
[0050]
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描 述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
[0051]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这 些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各 个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点 值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视 为在本文中具体公开。
[0052]
在以下实施例中,部分所使用的化学品的来源如下表1所示:
[0053]
表1
[0054]
[0055][0056]
实施例1化合物的制备
[0057]
化合物t-1的合成
[0058]
将二乙二醇单甲醚(58.65ml,500mmol)溶于160ml的四氢呋喃溶液 中,冰浴至0℃。将氢氧化钠(40g,1000mmol)溶于110ml水中,缓慢加 入。随后称取对甲苯磺酰氯(142.74g,750mmol)溶于110ml四氢呋喃溶 液中,使用恒压滴液漏斗缓慢滴加在混合液中。滴加完成后,将混合物在常 温下搅拌2h。使用tlc监测反应,反应结束后加入400ml水,分离出有机 层。然后使用1m的氢氧化钠溶液洗涤两次。收集有机相使用无水硫酸钠干 燥,并浓缩。得到t-1产物115.08g,产率约为84%。ms(esi)m/z理论值 c
12h18
o5s[m+na]
+
为278.33;测量值,298.6。1h nmr(400mhz,cdcl3)δ7.79 (d,j=8.0hz,2h),7.33(d,j=8.0hz,2h),4.16(t,j=4.9hz,2h),3.68(t,j= 4.9hz,2h),3.60-3.53(m,2h),3.50-3.44(m,2h),3.34(s,
3h),2.43(s,3h)。
[0059]
化合物t-2的合成
[0060]
将对苯二酚(14.64g,133.1mmol)、碳酸钾(73.47g,532.4mmol)加入 2l的圆底烧瓶中,加入1.1l的乙腈。混合物加热回流30min,然后冷却至 室温。将化合物t-1(72.95g,266.2mmol)溶于220ml乙腈中,缓慢滴加 至混合液中。加热回流搅拌72h。反应结束后冷却至室温,抽滤、收集上清 液浓缩。进行柱层析纯化(石油醚/乙酸乙酯90:10-50:50),以得到t-2产物 35.80g,产率约为85.6%。ms(esi)m/z理论值c
16h26
o6[m+na]
+
为337.37; 测量值,337.2。1h nmr(400mhz,cdcl3)δ6.82(s,1h),4.10-4.05(m,1h), 3.83-3.80(m,1h),3.72-3.69(m,1h),3.58-3.55(m,1h),3.38(s,2h)。
[0061]
化合物t-3的合成
[0062]
称取化合物t-2(25g,79.6mmol)、碘(22.3g,87.6mmol)以及碘酸钾 (6.8g,31.8mmol)放入1000ml的圆底烧瓶中。加入醋酸250ml、水25ml 以及浓硫酸3.3ml。加热回流48h。实验结束后,冷却至室温,加入10%硫 代硫酸钠和二氯甲烷洗涤除去多余的碘,然后使用饱和碳酸氢钠溶液洗涤两 次、水洗涤一次。收集有机相,加入无水硫酸钠干燥。浓缩有机相,滤渣使 用乙醇重结晶。得到t-3产物34.00g,产率为42.7%。ms(esi)m/z理论值 c
16h24
i2o6[m+na]
+
为589.17;测量值,589.87。1h nmr(400mhz,cdcl3)δ 7.28(s,2h),4.13(d,j=5.0hz,4h),3.90(t,j=4.9hz,4h),3.82-3.77(m,4h), 3.63-3.57(m,4h),3.42(s,6h)。
[0063]
化合物t-4的合成
[0064]
称取化合物t-3(5.66g,10mmol),四(三苯基膦)钯(1.2g,1.0mmol), 碘化亚铜(200mg,1.0mmol)和4-乙炔基苯胺(2.34g,20mmol)放入100ml 的两口烧瓶中,在无水无氧的条件下加入20ml的四氢呋喃和30ml的三乙 胺溶解。氮气保护下在45℃下反应24h。反应结束后,冷却至室温,旋转蒸 发除去溶剂。加入二氯甲烷和饱和碳酸氢钠溶液洗涤。有机相使用无水硫酸 钠干燥。浓缩后进行柱层析纯化,以得到t-4产物4.73g,产率为86.9%。 ms(esi)m/z理论值c
32h36
n2o6[m+na]
+
为567.64;测量值,568.8。1h nmr (400mhz,cdcl3)δ7.32(d,j=8.5hz,4h),6.99(s,2h),6.62(d,j=8.5hz, 4h),4.19(t,j=4.9hz,4h),3.91(t,j=4.9hz,4h),3.86(d,j=9.9hz,4h), 3.82-3.79(m,4h),3.57-3.51(m,4h),3.36(s,6h)。
[0065]
化合物t-5的合成
[0066]
化合物6-{4-[羟基-(4-甲氧基苯基)-苯基-甲基]苯氧基}己酸(2.1g, 5mmol)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(2.28g, 6mmol)、n,n-二异丙基乙胺(0.78g,6mmol)溶于一定量的二氯甲烷中,常 温搅拌30min。将混合液滴加在2.838g(5.0mmol)的化合物t-4的二氯甲烷 溶液中。温搅拌过夜。反应结束后,使用饱和碳酸氢钠溶液洗涤,有机相使 用无水硫酸钠干燥。浓缩后进行柱层析纯化,得到t-5产物3.42g,产率为 72.3%。ms(esi)m/z理论值c
58h62
n2o
10
[m-h]-为947.12;测量值,947.12。 1
h nmr(400mhz,cdcl3)δ8.03(s,1h),7.55(d,j=9.1hz,4h),7.35-7.33 (m,2h),7.31-7.28(m,5h),7.17(d,j=8.2hz,4h),7.02(d,j=2.2hz,2h), 6.82(d,j=9.1hz,4h),6.64(d,j=8.2hz,2h),5.32(s,2h),4.22(t,j=4.9hz, 4h),3.97-3.92(m,5h),3.82(d,j=3.9hz,4h),3.58-3.53(m,3h),3.38(d,j =2.9hz,4h),2.97(s,3h),2.90(s,4h),2.40(dd,j=8.7,6.2hz,2h),2.05(d,j =3.5hz,1h),1.84-1.79(m,4h),1.58-1.53(m,2h)。
[0067]
化合物t-6的合成
[0068]
添加化合物t-5(3.78g,4mmol)、无水二氯甲烷(20ml)和n,n-二异 丙基乙胺(1.56g,12mmol)到经干燥的圆底烧瓶中,冷却到0℃。在2ml 二氯甲烷中加入甲基丙烯酰氯(4mmol)溶液,通过加入漏斗缓慢搅拌。添 加后,反应瓶与氮气断开,通过干燥剂管与空气相连。混合物在室温下搅拌 了一夜。将反应物转移到分液漏斗中,使用10%的碳酸氢钠溶液和二氯甲烷 溶液洗涤。有机相用无水硫酸钠干燥,浓缩后进行柱层析纯化,得到t-6产 物3.40g,产率为83.7%。ms(esi)m/z理论值c
62h66
n2o
11
[m-h]-为1014.19; 测量值,1014.87。1h nmr(400mhz,cdcl3)δ7.61-7.44(m,9h),7.30(m,4h), 7.16(ddd,j=8.7,6.0,2.7hz,4h),7.03(s,2h),6.82(dd,j=8.9,7.4hz,4h), 5.81(s,1h),5.50(s,1h),4.21(t,j=4.8hz,4h),3.94(m,7h),3.85-3.74(m, 6h),3.54(dt,j=6.3,2.9hz,4h),3.36(d,j=2.7hz,6h),2.73(s,1h)2.40(t,j =7.4hz,2h),2.08(s,3h),1.81(m,4h),1.57(m,2h)。
[0069]
化合物t-7的合成
[0070]
取t-6(3.045g,3mmol)和n-4-乙酰基-2'-脱氧胞苷(0.81g,3mmol) 放置在两个圆底烧瓶中真空干燥过夜,在装有化合物t-6的烧瓶中加入乙酰 氯(4ml),在氮气下常温搅拌两个小时。真空抽干乙酰氯,用干的正己烷洗 涤,真空干燥1h。将n-4-乙酰基-2'-脱氧胞苷用5ml的吡啶溶解加入其中, 常温搅拌16h。反应结束后,旋转蒸发除去吡啶。浓缩液使用二氯甲烷和饱 和碳酸氢钠溶液洗涤,有机相用无水硫酸钠干燥,浓缩后进行柱层析纯化, 得到t-7产物1.50g,产率为39.4%。ms(esi)m/z理论值c
73h79
n5o
15
[m+h]
+
为1267.43;测量值,1267.80。1h nmr(400mhz,cdcl3)δ8.67(s,1h),8.16 (dd,j=7.5,2.6hz,1h),7.97(s,1h),7.64(t,j=8.0hz,3h),7.53-7.32(m, 6h),7.25(m,3h),7.24-7.08(m,4h),7.04-6.92(m,2h),6.80(ddd,j=13.5, 8.7,1.9hz,4h),6.21(t,j=5.8hz,1h),5.85(s,1h),5.47(s,1h),4.47(q,j= 4.8hz,1h),4.16(m,5h),3.97-3.84(m,6h),3.77(m,7h),3.51(m,4h),3.48
‑ꢀ
3.39(m,2h),3.33(m,6h),2.66(dt,j=12.5,5.7hz,1h),2.45(t,j=7.5hz, 2h),2.24(s,3h),2.18(m,1h),2.05(s,3h),1.77(h,j=6.3,5.6hz,4h),1.49 (m,2h)。
[0071]
化合物t-8的合成
[0072]
将化合物t-7(1.26g,1.0mmol)溶解于6ml无水二氯甲烷,加入5-苄 硫基四氮唑(307.2mg,1.6mmol)与2-氰乙基n,n,n',n'-四异丙基亚磷酰二胺 (600μl,1.5mmol)。反应在室温下搅拌1h后,用饱和碳酸氢钠溶液洗涤三 次,旋转蒸发除去有机相,用正己烷沉淀,以得到t-8产物1.34g,产率为 91.2%。ms(esi)m/z理论值c
82h96
n7o
16
p[m+h]
+
为1489.65;测量值,1489.01。 1
h nmr(400mhz,cdcl3)δ8.32-8.14(m,1h),7.66-7.42(m,9h),7.39(t,j =6.9hz,2h),7.32-7.23(m,6h),7.20-7.05(m,2h),7.02(d,j=2.3hz,2h), 6.83(ddd,j=10.1,7.5,5.1hz,4h),6.24(q,j=6.2hz,1h),5.81(s,1h),5.49 (s,1h),4.67-4.50(m,1h),4.20(d,j=4.8hz,4h),4.04-3.89(m,6h),3.79-85 (m,7h),3.66-3.48(m,8h),3.36(d,j=2.5hz,6h),2.72(ddd,j=26.0,13.0, 6.6hz,1h),2.54(q,j=7.2hz,4h),2.47-2.33(m,2h),2.20(s,3h),2.07(s, 3h),1.80(p,j=7.3hz,4h),1.65-1.56(m,2h),1.19-1.12(m,12h)。
31
p nmr (400mhz,cdcl3)δ149.41,149.38,148.76,148.73。
[0073]
化合物t-9的合成
[0074]
取t-6(3.045g,3mmol)和2'-脱氧胸苷(0.73g,3mmol)放置在两个圆 底烧瓶中真空干燥过夜,在装有化合物t-6的烧瓶中加入乙酰氯(4ml),在 氮气下常温搅拌两个小时。真空抽干乙酰氯,用干的正己烷洗涤,真空干燥 1h。将2'-脱氧胸苷用5ml的吡啶溶解加入
其中,常温搅拌16h。反应结束后, 旋转蒸发除去吡啶。浓缩液使用二氯甲烷和饱和碳酸氢钠溶液洗涤,有机相 用无水硫酸钠干燥,浓缩后进行柱层析纯化,得到t-9产物2.58g,产率69.0%。 ms(esi)m/z理论值c
72h78
n4nao
15
[m+na]
+
为1261.54;测量值,1261.60。1hnmr(400mhz,cdcl3)δ7.80(s,1h),7.63(d,j=3.7hz,2h),7.61(d,j=4.1 hz,2h),7.50(d,j=8.2hz,2h),7.45(d,j=8.3hz,2h),7.39(t,j=4.8hz, 3h),7.30-7.24(m,9h),7.02(s,2h),6.84-6.79(m,4h),6.41(t,j=6.8hz, 1h),5.84(s,1h),5.50(s,1h),4.59(d,j=9.0hz,1h),4.20(d,j=4.8hz,4h), 4.08(d,j=3.1hz,1h),3.97-3.90(m,7h),3.80(t,j=3.3hz,7h),3.57-3.53 (m,4h),3.44(d,j=6.8hz,1h),3.37(d,j=3.1hz,6h),2.44(q,j=7.2,6.5hz, 4h),2.08(s,3h),1.79(s,4h),1.56-1.51(m,2h),1.41(s,4h)。
[0075]
化合物t-10的合成
[0076]
将t-9(1.24g,1.0mmol)溶解于6ml无水二氯甲烷中,加入5-苄硫基 四氮唑(307.2mg,1.6mmol)与2-氰乙基n,n,n',n'-四异丙基亚磷酰二胺 (600μl,1.5mmol)。反应在室温下搅拌1h后,用饱和碳酸氢钠溶液洗涤三 次,旋转蒸发除去有机相,用正己烷沉淀,以得到t-10产物1.15g,产率为 79.8%。ms(esi)m/z理论值c
81h95
n6nao
16
p[m+na]
+
为1461.64;测量值, 1462.13。1h nmr(400mhz,cdcl3)δ7.78(d,j=5.4hz,1h),7.68(d,j=3.6 hz,2h),7.61-7.54(m,5h),7.48(t,j=7.7hz,4h),7.41(d,j=7.7hz,3h), 7.30-7.26(m,6h),7.04(d,j=1.9hz,2h),6.86-6.82(m,4h),6.45-6.39(m, 1h),5.83(s,1h),5.51(s,1h),4.69(dq,j=10.6,7.2,5.2hz,1h),4.22(t,j= 4.9hz,5h),3.99(d,j=6.7hz,2h),3.95-3.92(m,4h),3.81(d,j=2.2hz, 7h),3.60-3.55(m,7h),3.38(d,j=2.0hz,8h),2.78(dt,j=6.3,3.2hz,1h), 2.64(t,j=6.2hz,1h),2.44-2.38(m,4h),2.07(d,j=11.9hz,4h),1.81(t,j= 7.3hz,5h),1.40(d,j=3.6hz,4h),1.28(s,12h);
31
p nmr(400mhz,cdcl3) δ149.37,149.03,148.97,148.45。
[0077]
具体地,实施例1的整体合成路线如图1所示,其中,各步骤的条件如 下:a)naoh,thf,2h;b)对二苯酚,k2co3,乙腈,72h;c)i2,kio3, acoh,48h;d)pd(pph3)4,cui,4-乙炔基苯胺,thf,et3n,24h;e)6-{4-[羟 基-(4-甲氧基苯基)-苯基-甲基]苯氧基}己酸,diea,hatu,dcm,12h;f) diea,甲基丙烯酰氯,dcm,12h;g)乙酰氯,dc-ac,吡啶,16h;h)2
‑ꢀ
氰乙基n,n,n',n'-四异丙基亚磷酰二胺,btt,dcm,1h;以及i)乙酰氯, dt,吡啶,16h。
[0078]
实施例2纯化回收率的检测
[0079]
将一段25个碱基的寡核苷酸 (5
’‑
ho-cacactctttccctacacgacgctc-oh-3’,seq id no:1)用 于验证本发明的捕获和释放策略在寡核苷酸纯化中的应用。寡核苷酸序列 (cacactctttccctacacgacgctc)的位置信息编辑完毕之后,上载 于自动化dr.oligo48dna合成仪。单体a、c、g、t亚磷酰胺单体溶解于 二氯甲烷,浓度为0.06m;分别置于合成仪单独的对应合成通道后,合成规 模为200nmol或者1000nmol,模式为dmtr on,固相合成及切割脱保护的方 法同常规的寡核苷酸固相合成。经过切割脱除碱基保护基团后的粗品经质谱 分析和hplc分析,hplc采用waters xbridge oligonucleotide beh c18色 谱柱,型号为186003953。粗品分析hplc梯度洗脱条件为0-6min,乙腈浓 度为5%-50%;6.01-8min,乙腈浓度为88%;8.01-10min,乙腈浓度为5%。 如图2所示,寡核苷酸的保留时间为4.842min,未经纯化的寡核苷酸纯度为 83.86%,显示每步的缩合效率平均为99.27%。
[0080]
作为试验组,用单体t-8替换普通的c单体,将化合物t-8溶解于乙腈, 使其浓度为
0.10m,并将t-8单体连接在一个长度为24个碱基的寡核苷酸的 5’端(5
’‑
ho-acactctttccctacacgacgctc-oh-3’,seq id no:2, 目标结构如图3所示)。化合物t-8偶联时间为180s
×
6次,偶联总时间为 18min。合成结束后,加入1.5ml的氨水,在55℃下氨解15h后,浓缩抽干 得到粗品。取200nmol的粗寡核苷酸溶于50μl水,振荡使其充分溶解。加 入聚合试剂(24μl,3.7m的n,n-二甲基丙烯酰胺和0.18m的n,n
′‑
亚甲基双 丙烯酰胺),5μl的5%过硫酸铵,5μl的0.66m四甲基乙二胺。振荡使其混 合均匀。常温静置15min后出现凝胶,1h后聚合反应结束。将凝胶状聚合物 切成碎片,加入250μl 5%三乙胺水溶液,等待3min后,除去溶液,此步骤 重复5次。最后加入250μl超纯水洗去残留的三乙胺溶液。然后向凝胶中加 入能覆盖凝胶最低量的80%醋酸溶液(约100μl),5min后,收集80%醋酸 溶液(acoh),重复3次。然后向凝胶中加入100μl水,3min后收集水溶 液,重复5次。将收集的醋酸溶液和水溶液合并后真空浓缩。向浓缩后的寡 核苷酸中加入100μl浓氨水,短暂振荡后放入85℃下15min。冷却至室温后 加入900μl正丁醇,振荡1min后,放在高速离心机中,在14000rpm下离心 15min。用移液器小心移除上清液,浓缩除去多余的正丁醇。留下的白色固 体为纯化后的寡核苷酸。产物经hplc分析,采用岛津shim-pack gist高纯 硅胶色谱柱,型号为227-30011-03。纯品分析hplc梯度洗脱条件为0-6min, 乙腈浓度为5%-50%;6.01-8min,乙腈浓度为88%;8.01-10min,乙腈浓度 为5%。如图4所示,经过纯化后得到的产品,保留时间为2.477min;其分 子量由esi质谱确认无误(图5),ms(esi)m/z理论值7458.87,测量值为7458。 回收量为53.37nmol,回收率为26.7%,较已报道的基于捕获和释放策略的试 剂回收率提高了53%,寡核苷酸纯度相当为99.40%。
[0081]
用于1000nmol合成规模的纯化时,取1000nmol的粗寡核苷酸溶于100μl 水,振荡使其充分溶解,加入聚合试剂(60μl,3.7m n,n-二甲基丙烯酰胺 和0.18m n,n
′‑
亚甲基双丙烯酰胺),5μl的5%过硫酸铵以及5μl的0.66m 四甲基乙二胺,振荡使其混合均匀。常温静置15min后出现凝胶状态,1h后 聚合反应结束。将凝胶状聚合物切成碎片,加入500μl 5%三乙胺溶液,等待 3min后,除去溶液,此步骤重复5次。最后加入500超纯水洗去残留的三 乙胺溶液。然后向凝胶中加入能覆盖凝胶最低量的80%醋酸溶液(约300μl), 5min后,收集80%醋酸溶液,重复3次。然后向凝胶中加入300μl水,3min 后收集水溶液,重复5次。将收集的醋酸溶液和水溶液合并后真空浓缩。向 浓缩残渣中加入200μl浓氨水,短暂振荡后放入85℃下15min。冷却至室温 后加入1800μl正丁醇,振荡1min后,放在高速离心机中,在14000rpm下 离心15min。用移液器小心移除上清液,浓缩除去多余的正丁醇。留下的白 色固体为纯化后的寡核苷酸。产物经hplc分析,采用岛津shim-pack gist 高纯硅胶色谱柱,型号227-30011-03。纯品分析hplc梯度洗脱条件为 0-6min,乙腈浓度为5%-50%;6.01-8min,乙腈浓度为88%;8.01-10min,乙 腈浓度为5%。如图6所示,经过纯化后得到干净的产品,保留时间为 2.462min;其分子量由esi质谱确认无误(图7)。ms(esi)m/z理论值7458.87, 测量值为7458。回收量为227.7nmol,回收率为22.77%,较已有的基于捕获 和释放策略的试剂回收率提高了30%,寡核苷酸纯度为99.78%。
[0082]
作为试验组,采用t-10作为纯化剂,其用于纯化25个碱基长的寡核苷 酸(5
’‑
ho-tacactctttccctacacgacgctc-oh-3’,seq id no:3), 200nmol合成规模,回收量为53.8nmol,回收率为26.90%,较已有的基于捕 获和释放策略的试剂回收率提高了54%,寡核苷酸纯度为99.26%(图8),分 子量由esi质谱确认无误(图9),ms(esi)m/z理论值
7472.87,测量值为7473; 1000nmol的回收量为262nmol,回收率为26.20%,较已有的基于捕获和释放 策略的试剂回收率提高了50%,寡核苷酸纯度为99.13%(图10),分子量由 esi质谱确认无误(图11),ms(esi)m/z理论值7472.87,测量值为7473。
[0083]
实施例3验证寡核苷酸的下游应用
[0084]
为了验证使用本发明的纯化方法纯化后的寡核苷酸对下游应用无影响, 选择聚合酶链式反应(pcr)和即时聚合酶链式反应(qpcr)两种应用进行 测试。经本发明纯化后的寡核苷酸与经过hplc纯化的寡核苷酸分别用于 pcr和qpcr。
[0085]
将经本发明和hplc两种纯化方法纯化的正向寡核苷酸引物和经hplc 纯化的反向寡核苷酸引物的浓度分别配制为10μm,模板浓度为1.5pg/μl。 在pcr管中分别加入2μl正向引物、2μl反向引物、1μl模板(引物及模板 序列如下表2所示)、25μl kapa sybr fast qpcr master mix(2x)以及 20μl水,放入pcr仪中在设定程序(95℃3min,98℃20s,60℃30s,72℃ 30s,72℃5min)中扩增30个循环。实验结束后,每管pcr分别取4μl使 用2.5%琼脂糖胶进行分析,结果如图12所示,显示两种方式纯化的正向引 物用于pcr反应后,结果无差别。
[0086]
表2
[0087][0088]
用于qpcr反应时,将正向引物和反向引物的浓度分别配制为10μm, 模板浓度为1.5pg/μl。在pcr管中分别加入0.6μl正向引物、0.6μl反向引 物、1μl模板、10μl kapa sybr fast qpcr master mix(2x)、0.6μl roxlow溶液以及7.4μl水,放入qpcr仪中在设定程序(95℃3min,98℃20s, 60℃30s,72℃30s,95℃15s,60℃1min,95℃1s)中扩增40个循环,平 行三组。扩增曲线图如图13所示,ct值示于表3中。每管pcr中分别取4μl 使用2.5%琼脂糖胶进行分析。显示两种方式纯化的正向引物用于qpcr反应 后,ct值的结果无差别,胶图(图14)都显示正确寡核苷酸大小。
[0089]
表3
[0090]
[0091]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实 施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方 案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0092]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征, 在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的 重复,本发明对各种可能的组合方式不再另行说明。
[0093]
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其 不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1