一种头孢布烯反式异构体的制备方法与流程
1.本发明涉及药物相关杂质合成技术领域,尤其涉及一种头孢布烯反式异构体的制备方法。
背景技术:
2.头孢布烯(ceftibuten),化学名为[6r-[(6α,7β(z)]]-7-[[2-(2-氨基-4-噻唑基)-4-羧基-1-氧-2-丁烯基]氨基]-8-氧-5-硫杂-1-氮杂二环[4.2.0]辛-2-烯-2-羧酸,其化学结构如下式所示:
[0003][0004]
头孢布烯(ceftibuten)是由日本盐野义公司研制的第三代广谱口服头孢菌素,对大多数革兰氏阴性杆菌及部分阳性球菌有较强的抗菌作用,对质粒介导的β-内酰胺酶高度稳定,且具有抗生素后效应,具有抗菌谱广、抗菌活性强、生物利用度高等特点,用于治疗由敏感菌株引起的各种感染,包括上呼吸道感染、下呼吸道感染、泌尿系统感染、肠炎和胃肠炎等。
[0005]
杂质是活性药物成分(api)或药品制剂中不希望存在的化学成分,它们可能来源于api或药品制剂的生产过程,也可能来源于原料药的储存过程;它们可能是已知、未知、挥发或不挥发的化合物,其来源包括原料、中间体、意外副产物和降解产物;它们也可能来自于对映体间的消旋或污染。所有这些情况下产生的杂质都有可能导致不良的生物活性或毒性。原料药中各种杂质的含量决定了最终成品药物的安全性。因此,杂质的鉴定、定量、定性和控制已成为药物开发过程的关键组成部分。
[0006]
头孢布烯反式异构体作为一种在合成、储存、运输的过程中均会引入的一种杂质,是质量控制中需要研究的重要杂质。但是,目前该杂质缺少较为成熟的获取方法。
技术实现要素:
[0007]
有鉴于此,本发明提供了一种头孢布烯反式异构体的制备方法。本发明提供的制备方法简单高效,可用于提供大量高品质的杂质对照品,对头孢布烯的质量控制起到积极的作用。
[0008]
为了实现上述发明目的,本发明提供以下技术方案:
[0009]
一种头孢布烯反式异构体的制备方法,包括以下步骤:
[0010]
将双保护头孢布烯、lewis酸和有机溶剂混合进行脱保护反应,反应完成后使用盐酸溶液将反应淬灭,之后进行静置分层,得到水层;
[0011]
在-10~10℃条件下,将所述水层的ph值调节至2.0~3.5,然后在0~20℃下进行
转晶,之后依次进行固液分离、干燥和纯化,得到头孢布烯反式异构体。
[0012]
优选的,所述lewis酸包括无水三氯化铝、四氯化钛和无水三氯化铁中的一种或几种。
[0013]
优选的,所述lewis酸与双保护头孢布烯的摩尔比为1:(3~5.3)。
[0014]
优选的,所述有机溶剂包括苯甲醚、二氯甲烷和三氯甲烷中的一种或几种。
[0015]
优选的,所述双保护头孢布烯、lewis酸和有机溶剂混合的方法具体为:将双保护头孢布烯溶解于部分有机溶剂中,得到双保护头孢布烯溶液,将lewis酸溶解于剩余部分有机溶剂中,得到lewis酸溶液,然后将所述双保护头孢布烯溶液和lewis酸溶液混合。
[0016]
优选的,所述脱保护反应的温度为15~30℃,时间为50~80min。
[0017]
优选的,调节所述水层ph值用试剂为氢氧化钠水溶液。
[0018]
优选的,调节所述水层的ph值之前,还包括将所述水层进行萃洗,所述萃洗用试剂为二氯甲烷。
[0019]
优选的,所述转晶的时间为2~4h。
[0020]
优选的,所述纯化的方法为柱层析纯化,所述柱层析纯化用洗脱试剂为乙腈-水混合溶剂。
[0021]
本发明提供了一种头孢布烯反式异构体的制备方法,包括以下步骤:将双保护头孢布烯、lewis酸和有机溶剂混合进行脱保护反应,反应完成后使用盐酸溶液将反应淬灭,之后进行静置分层,得到水层;在-10~10℃条件下,将所述水层的ph值调节至2.0~3.5,然后在0~20℃下进行转晶,之后依次进行固液分离、干燥和纯化,得到头孢布烯反式异构体。本发明首先对双保护头孢布烯脱保护,然后在一定温度和ph值条件下进行转晶,成功制备了头孢布烯反式异构体。本发明提供的方法简单高效,成本低,且产品纯度高、收率高,可用于提供大量高品质的杂质对照品,为药品安全使用提供理论依据,对头孢布烯生产过程的质量控制提供有效的数据支持。实施例结果表明,本发明制备的头孢布烯反式异构体纯度为95%以上,摩尔收率为60%以上。
附图说明
[0022]
图1为实施例1制备的孢布烯反式异构体的hplc图谱。
具体实施方式
[0023]
本发明提供了一种头孢布烯反式异构体的制备方法,包括以下步骤:
[0024]
将双保护头孢布烯、lewis酸和有机溶剂混合进行脱保护反应,反应完成后使用盐酸溶液将反应淬灭,之后进行静置分层,得到水层;
[0025]
在-10~10℃条件下,将所述水层的ph值调节至2.0~3.5,然后在0~20℃下进行转晶,之后依次进行固液分离、干燥和纯化,得到头孢布烯反式异构体。
[0026]
本发明将双保护头孢布烯、lewis酸和有机溶剂混合进行脱保护反应。在本发明中,所述lewis酸优选包括无水三氯化铝、四氯化钛和无水三氯化铁中的一种或几种;所述lewis酸与双保护头孢布烯的摩尔比优选为1:(3~5.3),更优选为1:(3.5~4.5);所述有机溶剂优选包括苯甲醚、二氯甲烷和三氯甲烷中的一种或几种;本发明对所述有机溶剂的用量没有特殊要求,能够保证脱保护反应的顺利进行即可。
[0027]
在本发明中,所述双保护头孢布烯、lewis酸和有机溶剂混合的方法具体为:将双保护头孢布烯溶解于部分有机溶剂中,得到双保护头孢布烯溶液,将lewis酸溶解于剩余部分有机溶剂中,得到lewis酸溶液,然后将所述双保护头孢布烯溶液和lewis酸溶液混合。在本发明的具体实施例中,所述双保护头孢布烯溶液的浓度优选为0.15~0.25mol/l,所述lewis酸溶液的浓度优选为0.70~1.2mol/l。
[0028]
在本发明中,所述脱保护反应的温度优选为15~30℃,更优选为20~25℃,时间优选为50~80min,更优选为60~70min。
[0029]
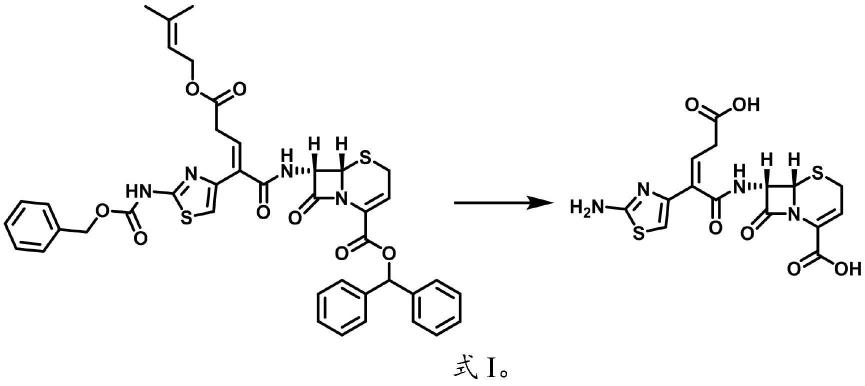
在本发明中,所述脱保护反应的反应方程式如式i所示,脱保护反应所得产物为头孢布烯顺反异构体的混合物。
[0030][0031]
脱保护反应完成后,本发明采用盐酸溶液将反应淬灭,之后进行静置分层,得到水层。在本发明中,所述盐酸溶液的质量浓度优选为2~5%,更优选为3~4%,所述脱保护反应所得反应液与盐酸溶液的用量比优选为200~250ml:100~250g,更优选为230~250ml:120~230g。本发明对所述静置分层的方法没有特殊要求,采用本领域技术人员熟知的方法即可,静置分层后,脱保护产物进入水层中。
[0032]
得到水层后,本发明优选在-10~10℃条件下,将所述水层的ph值调节至2.0~3.5,然后在0~20℃下进行转晶。在本发明中,优选在调节所述水层的ph值之前,先对所述水层进行萃洗,所述萃洗用试剂优选为二氯甲烷,所述萃洗的次数优选为3次;萃洗完成后,优选将萃洗后的水层降温至-10~10℃,更优选降温至-10~-5℃、-5~0℃、0~5℃或5~10℃,之后再调节所述水层的ph值,调节所述水层ph值用试剂优选为氢氧化钠水溶液,所述氢氧化钠水溶液的质量分数优选为20~30%,更优选为25~28%;在降温和调节ph值的过程中,结晶产物从水相中析出。
[0033]
调节水层的ph值后,本发明在0~20℃下进行转晶,优选在0~5℃、5~10℃或15~20℃下进行转晶;所述转晶的时间优选为2~4h,更优选为2.5~3.5h。本发明在上述温度和ph值条件下进行转晶,使头孢布烯顺式异构体向反式异构体转化。
[0034]
转晶完成后,本发明依次进行固液分离、干燥和纯化,得到头孢布烯反式异构体。在本发明中,所述固液分离的方法优选为过滤;所述干燥的温度优选为40℃,本发明对所述干燥的时间没有特殊要求,以充分干燥为准。在本发明中,所述纯化的方法优选为柱层析纯化,所述柱层析纯化用洗脱试剂优选为乙腈-水混合溶剂,所述混合溶剂中乙腈和水的体积
比优选为1:(1~10),更优选为1:(2~5),进一步优选为1:(3~4)。
[0035]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0036]
实施例1
[0037]
将双保护头孢布烯18g(0.023mol)投入100ml苯甲醚中,搅拌溶清;将无水三氯化铝15g(0.112mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于20~25℃下反应60min,反应结束后使用180g的3%盐酸淬灭,水层用50g二氯甲烷萃洗3次,于0~5℃下用28%氢氧化钠水溶液调ph至2.2
±
0.2,在10~15℃下转晶2.5h,过滤,得到粗品,经过柱层析纯化,洗脱体系为乙腈:水=1:5,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得7.1g(摩尔收率68.8%),经hplc检测纯度98.6%。
[0038]
纯化所得头孢布烯反式异构体的hplc图谱如图1所示,hplc谱图数据如表1所示。
[0039]
表1 hplc谱图数据
[0040][0041][0042]
实施例2
[0043]
将双保护头孢布烯18g(0.023mol)投入150ml二氯甲烷中,搅拌溶清;将无水三氯化铁12.2g(0.075mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加
入其中,于15~20℃下反应70min,反应结束后使用120g的5%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-10~-5℃下用28%氢氧化钠水溶液调ph至2.5
±
0.2,在10~15℃下转晶3h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:1,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.5g(摩尔收率63.0%),经hplc检测纯度95.3%。
[0044]
实施例3
[0045]
将双保护头孢布烯18g(0.023mol)投入120ml三氯甲烷中,搅拌溶清;将无水三氯化铁15.9g(0.098mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于18~22℃下反应80min,反应结束后使用140g的4%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-5~0℃下用28%氢氧化钠水溶液调ph至2.6
±
0.2,在15℃~20℃下转晶3.5h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:2,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.2g(摩尔收率60.0%),hplc检测纯度97.4%。
[0046]
实施例4
[0047]
将双保护头孢布烯18g(0.023mol)投入150ml二氯甲烷中,搅拌溶清;将四氯化钛13.3g(0.07mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于25~30℃下反应55min,反应结束后使用250g的2%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于0~5℃下用28%氢氧化钠水溶液调ph至2.8
±
0.2,在10~15℃下转晶2h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:1,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.9g(摩尔收率66.9%),经hplc检测纯度98.1%。
[0048]
实施例5
[0049]
将双保护头孢布烯18g(0.023mol)投入100ml苯甲醚中,搅拌溶清;将无水四氯化钛13.3g(0.07mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于20~25℃下反应60min,反应结束后使用120g的5%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于5~10℃下用28%氢氧化钠水溶液调ph至3.3
±
0.2,在5~10℃下转晶4h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:3,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得7.3g(摩尔收率70.7%),经hplc检测纯度95.6%。
[0050]
实施例6
[0051]
将双保护头孢布烯18g(0.023mol)投入150ml二氯甲烷中,搅拌溶清;将无水三氯化铝16.0g(0.12mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于10~15℃下反应70min,反应结束后使用180g的3%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-5~0℃下用28%氢氧化钠水溶液调ph至3.0
±
0.2,在15~20℃下转晶3.5h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:3,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.6g(摩尔收率64.0%),经hplc检测纯度96.9%。
[0052]
实施例7
[0053]
将双保护头孢布烯18g(0.023mol)投入120ml三氯甲烷中,搅拌溶清;将无水三氯化铝15.0g(0.112mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于15~20℃下反应60min,反应结束后使用140g的4%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-10~-5℃下用28%氢氧化钠水溶液调ph至2.8
±
0.2,在10~15℃下转晶3.5h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:5,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.4g(摩尔收率62.0%),经hplc检测纯度96.0%。
[0054]
实施例8
[0055]
将双保护头孢布烯18g(0.023mol)投入120ml三氯甲烷中,搅拌溶清;将无水三氯化铝14.6g(0.11mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于15~20℃下反应65min,反应结束后使用180g的3%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-10~-5℃下用28%氢氧化钠水溶液调ph至2.2
±
0.2,在10~15℃下转晶2.5h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:2,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得6.9g(摩尔收率66.9%),经hplc检测纯度95.3%。
[0056]
实施例9
[0057]
将双保护头孢布烯18g(0.023mol)投入150ml二氯甲烷中,搅拌溶清;将无水三氯化铝16.0g(0.12mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于20~25℃下反应60min,反应结束后使用140g的4%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于0~5℃下用28%氢氧化钠水溶液调ph至3.3
±
0.2,在15~20℃下转晶4h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:2,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得7.9g(摩尔收率76.6%),经hplc检测纯度96.8%。
[0058]
实施例10
[0059]
将双保护头孢布烯18g(0.023mol)投入100ml苯甲醚中,搅拌溶清;将四氯化钛17.3g(0.09mol)投入100ml苯甲醚中,搅拌溶清后,将溶清的双保护头孢布烯溶液加入其中,于20~25℃下反应60min,反应结束后使用120g的5%盐酸溶液淬灭,水层用50g二氯甲烷萃洗3次,于-10~-5℃下用28%氢氧化钠水溶液调ph至2.5
±
0.2,在15~20℃下转晶2.5h,过滤,得到粗品,经柱层析纯化,洗脱体系为乙腈:水=1:3,纯化并浓缩洗脱剂得头孢布烯反式异构体,40℃烘干得7.0g(摩尔收率67.8%),经hplc检测纯度95.4%。
[0060]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1