卡巴拉汀关键手性中间体的纯化方法与外消旋化回收利用
1.本发明属于药物合成领域,更具体地,涉及一种卡巴拉汀关键手性中间体的纯化方法与外消旋化回收利用,能够实现卡巴拉汀中间体的手性拆分和外消旋化回收。
背景技术:
2.阿尔兹海默症(alzheimer disease,ad),又称为老年痴呆症,是一种渐进性的神经功能退化性失调症,其主要临床表现为记忆障、失语、失认、执行功能障碍等。研究证实ad患者基底前脑的胆碱能神经细胞明显缺失,特别是在新皮层、海马体和杏仁核中。且胆碱能的损伤程度与ad的严重程度成正相关。所以目前用来ad的治疗药物也是针对乙酰胆碱系统来改善患者的症状。
3.卡巴拉汀,结构式如下所示:
[0004][0005]
其化学名为(s)-n-甲基-n-乙基-3-[1-(二甲氨基)乙基]-氨基甲酸苯酯,是毒扁豆碱的衍生物。不仅对脑部乙酰胆碱酯酶的选择性和作用效果好,而且副作用小。作为新一代乙酰胆碱酯酶抑制剂的代表,是一种假性不可逆乙酰胆碱酯抑制剂。主要用于轻中度阿尔兹海默症的对症治疗,能够明显延缓痴呆进程,改善患者的临床症状、认知功能和精神症状,具有良好依从性。目前合成卡巴拉汀的方法有三种:不对称合成法、手性酸拆分法和生物酶拆分法。不对称合成法和生物酶拆分法成本高、条件复杂且不适用于大规模生产。从成本及可行性的角度考虑,利用手性酸拆分不仅原料易得、条件温和,而且易于工业化放大。但是该方法也存在两个问题,一是拆分产率未达到最大值,能否找到更高效的拆分试剂。二是r构型对映体作为副产物能否回收利用,避免原料浪费,降低工业成本。
[0006]
中国专利文献cn101580482a采用从中间体3-[1-(二甲氨基)乙基]苯酚用乙醇和乙酸乙酯混合溶液重结晶拆分,得到(s)-3-[1-(二甲氨基)乙基]苯酚后,收率为27%。
[0007]
中国专利文献cn113461554a采用d-(+)-樟脑磺酸对中间体3-[1-(二甲氨基)乙基]苯酚进行多次重结晶拆分,收率为35.2%。
技术实现要素:
[0008]
针对现有技术的以上缺陷或改进需求,本发明的目的在于提供一种卡巴拉汀关键手性中间体的纯化方法与外消旋化回收利用,针对消旋体1-(3-甲氧基苯基)乙胺这一卡巴拉汀中间体,通过使用苯甲酰基-l-苯丙氨酸作、苯甲酰基-l-苯甘氨酸作为拆分试剂,能够有效实现纯化;并且,本发明还通过特定工艺流程设计的回收步骤,能够有效利用上述纯化工艺得到的副产物(即,(r)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯丙氨酸盐、(r)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯甘氨酸盐),最终得到消旋体1-(3-甲氧基苯基)乙胺,实现消
旋体1-(3-甲氧基苯基)乙胺的回收。与其他卡巴拉汀中间体的制备方法相比,本发明制备的卡巴拉汀中间体((s)-1-(3-甲氧基苯基)乙胺)产率高、效果好,而且避免了合成时产出的(r)-1-(3-甲氧基苯基)乙胺的浪费,有利于大规模工业化生产,能够有效解决现有合成方法产率低、成本高、拆分副产物未回收等问题。
[0009]
为实现上述目的,按照本发明的一个方面,提供了一种卡巴拉汀中间体的纯化方法,该卡巴拉汀中间体具体为消旋体1-(3-甲氧基苯基)乙胺,其特征在于,纯化方法包括以下步骤:
[0010]
(1)以消旋体1-(3-甲氧基苯基)乙胺为原料,将消旋体1-(3-甲氧基苯基)乙胺溶解于乙腈中,使用苯甲酰基-l-苯丙氨酸作或苯甲酰基-l-苯甘氨酸为拆分试剂,在加热搅拌的条件下进行拆分反应,反应后冷却,接着过滤;记滤渣对应的产物为中间体1,则:
[0011]
当拆分试剂为苯甲酰基-l-苯丙氨酸时,过滤得到的滤渣即为(s)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯丙氨酸盐,对应中间体1;
[0012]
当拆分试剂为苯甲酰基-l-苯甘氨酸时,过滤得到的滤渣即为(s)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯甘氨酸盐,对应中间体1;
[0013]
(2)将所述中间体1使用碱性溶液进行解离,即可获得(s)-1-(3-甲氧基苯基)乙胺。
[0014]
作为本发明的进一步优选,所述步骤(1)中,所述消旋体1-(3-甲氧基苯基)乙胺的质量与所述乙腈的体积之比为1:5至1:20g/ml;
[0015]
所述加热搅拌是在50℃至90℃的加热温度下进行的,更优选是在80℃的加热温度下进行的;
[0016]
所述步骤(2)中,所述碱性溶液选自氢氧化钠溶液、氢氧化钾溶液、碳酸钠溶液;
[0017]
优选的,所述步骤(2)具体是:首先将所述中间体1使用碱性水溶液解离,二氯甲烷萃取,合并有机溶剂,干燥后过滤旋干即可得到(s)-1-(3-甲氧基苯基)乙胺。
[0018]
按照本发明的另一方面,本发明提供了一种卡巴拉汀中间体的回收方法,该卡巴拉汀中间体具体为消旋体1-(3-甲氧基苯基)乙胺,其特征在于,包括以下步骤:
[0019]
(s1)以(r)-1-(3-甲氧基苯基)乙胺为待回收原料,记该(r)-1-(3-甲氧基苯基)乙胺为中间体3;将所述中间体3溶于第一溶剂中,并加入间甲氧基苯乙酮和钛酸四异丙酯,加热回流进行反应;反应结束后,冷却、蒸发除去第一溶剂得到反应浓缩液;
[0020]
(s2)将所述步骤(s1)中获得的反应浓缩液溶于第二溶剂中,并加入叔丁醇钾和二甲亚砜,加热回流进行反应;反应结束后,冷却,通过后处理得到间甲氧基苯乙酮和外消旋1-(3-甲氧基苯基)乙胺,能够实现外消旋1-(3-甲氧基苯基)乙胺的回收。
[0021]
按照本发明的又一方面,本发明提供了一种卡巴拉汀中间体的纯化及回收方法,该卡巴拉汀中间体具体为消旋体1-(3-甲氧基苯基)乙胺,其特征在于,纯化及回收方法包括以下步骤:
[0022]
(1)以消旋体1-(3-甲氧基苯基)乙胺为原料,将消旋体1-(3-甲氧基苯基)乙胺溶解于乙腈中,使用苯甲酰基-l-苯丙氨酸作或苯甲酰基-l-苯甘氨酸为拆分试剂,在加热搅拌的条件下进行拆分反应,反应后冷却,接着过滤;记滤渣对应的产物为中间体1,滤液对应的产物为中间体2,则:
[0023]
当拆分试剂为苯甲酰基-l-苯丙氨酸时,过滤得到的滤渣即为(s)-1-(3-甲氧基苯
基)乙胺苯甲酰基-l-苯丙氨酸盐,对应中间体1;滤液蒸发即可获得(r)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯丙氨酸盐,对应中间体2;
[0024]
当拆分试剂为苯甲酰基-l-苯甘氨酸时,过滤得到的滤渣即为(s)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯甘氨酸盐,对应中间体1;滤液蒸发即可获得(r)-1-(3-甲氧基苯基)乙胺苯甲酰基-l-苯甘氨酸盐,对应中间体2;
[0025]
(2)将所述中间体1使用碱性溶液进行解离,即可获得(s)-1-(3-甲氧基苯基)乙胺;
[0026]
(3)将所述中间体2使用碱性溶液进行解离,获得(r)-1-(3-甲氧基苯基)乙胺,记该(r)-1-(3-甲氧基苯基)乙胺为中间体3;接着,将所述中间体3溶于第一溶剂中,并加入间甲氧基苯乙酮和钛酸四异丙酯,加热回流进行反应;反应结束后,冷却、蒸发除去第一溶剂得到反应浓缩液;
[0027]
(4)将所述步骤(3)中获得的反应浓缩液溶于第二溶剂中,并加入叔丁醇钾和二甲亚砜,加热回流进行反应;反应结束后,冷却,通过后处理得到间甲氧基苯乙酮和外消旋1-(3-甲氧基苯基)乙胺,能够实现外消旋1-(3-甲氧基苯基)乙胺的回收。
[0028]
作为本发明的进一步优选,所述步骤(1)中,所述滤液蒸发具体是对滤液进行旋转蒸发;
[0029]
所述步骤(3)中,所述蒸发具体为旋转蒸发。
[0030]
作为本发明的进一步优选,所述步骤(2)和所述步骤(3)中,所述碱性溶液独立的选自:氢氧化钠溶液、氢氧化钾溶液、碳酸钠溶液。
[0031]
作为本发明的进一步优选,所述步骤(s1)、所述步骤(3)中,所述第一溶剂选自:甲苯、氯苯、对二甲苯;
[0032]
所述中间体3的质量与所述第一溶剂的体积之比为1:1至1:20g/ml;
[0033]
所述中间体3与所述间甲氧基苯乙酮的质量比为1:0.2至1:2;
[0034]
所述中间体3与所述钛酸四异丙酯的质量比为1:0.6至1:6;
[0035]
所述加热回流所采用的反应温度为90℃至140℃,反应时间为1h至48h。
[0036]
作为本发明的进一步优选,所述步骤(s2)、所述步骤(4)中,所述第二溶剂选自:叔丁醇、叔戊醇、乙二醇;
[0037]
所述加热回流所采用的反应温度为80℃至180℃,反应时间为1h至24h;
[0038]
所述反应浓缩液所对应采用的所述中间体3的质量与所述第二溶剂的体积之比为1:1至1:20g/ml;
[0039]
所述反应浓缩液所对应采用的所述中间体3与所述叔丁醇钾的质量比为1:0.45至1:4.5;
[0040]
所述反应浓缩液所对应采用的所述中间体3与所述二甲亚砜的质量比为1:0.25至1:3。
[0041]
作为本发明的进一步优选,所述步骤(2)具体是:首先将所述中间体1使用碱性水溶液解离,二氯甲烷萃取,合并有机溶剂,干燥后过滤旋干即可得到(s)-1-(3-甲氧基苯基)乙胺。
[0042]
作为本发明的进一步优选,所述步骤(3)中,将所述中间体2使用碱性水溶液进行解离,获得(r)-1-(3-甲氧基苯基)乙胺,具体是:首先将所述中间体2使用碱性水溶液解离,
(3-甲氧基苯基)乙基)氨基甲酸叔丁酯的hplc谱图。
[0053]
图5为实施例6中回收的1-(3-甲氧基苯基)乙胺与二碳酸二叔丁酯反应生成的(1-(3-甲氧基苯基)乙基)氨基甲酸叔丁酯的hplc谱图。
[0054]
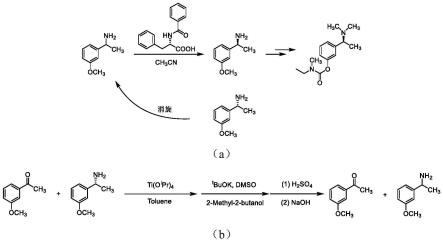
图6为卡巴拉汀关键中间体1-(3-甲氧基苯基)乙胺拆分和回收的反应流程;其中,图6中的(a)对应外消旋1-(3-甲氧基苯基)乙胺的拆分,图6中的(b)对应拆分副产物(r)-1-(3-甲氧基苯基)乙胺的外消旋化回收。
具体实施方式
[0055]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
[0056]
实施例1(s)-1-(3-甲氧基苯基)乙胺
[0057]
合成路线如下:
[0058][0059]
本实施例所采用的具体步骤如下:
[0060]
在室温下,向500ml圆底烧瓶中依次加入乙腈(200ml)、1-(3-甲氧基苯基)乙胺(15.1g,100mmol)和苯甲酰基-l-苯丙氨酸(26.9g,100mmol),80℃搅拌至澄清后再搅拌1h。关闭加热并停止搅拌,自然冷却至室温,放置数小时,直至不再析出白色不溶物。将反应液用多孔漏斗抽滤并用乙腈(50ml)洗涤固体,滤饼50℃鼓风干燥,得到(s)-1-(3-甲氧基苯基)乙胺的苯甲酰基-l-苯丙氨酸盐(中间体1,16.4g,39mmol)。
[0061]
滤液旋转蒸发得到(r)-1-(3-甲氧基苯基)乙胺的苯甲酰基-l-苯丙氨酸盐(中间体2,25.6g,61mmol),该中间体2可以进一步投入回收步骤(详见后文,尤其是实施例3-7)。
[0062]
再将中间体1溶于氢氧化钠溶液(2m,150ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用30g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(s)-1-(3-甲氧基苯基)乙胺(5.9g,39mmol)。
[0063]
拆分总收率为39%。
[0064]
产物(s)-1-(3-甲氧基苯基)乙胺的核磁检测结果为:
[0065]1h nmr(400mhz,cdcl3)δ7.25
–
7.20(m,1h),6.95
–
6.89(m,2h),6.80
–
6.75(m,1h),4.09(q,j=6.6hz,1h),3.80(s,3h),1.39(d,j=6.6hz,3h).
[0066]
(s)-1-(3-甲氧基苯基)乙胺衍生的(s)-(1-(3-甲氧基苯基)乙基)氨基甲酸叔丁酯,其ee值是通过高效液相色谱仪检测的,测试条件:as-h手性柱、流动相为正己烷:异丙醇
甲氧基苯基)乙胺,能够实现外消旋1-(3-甲氧基苯基)乙胺的回收(回收得到的间甲氧基苯乙酮,同样也可以重复利用)。
[0082]
当然,对于其他拆分方法获得的r型副产物,同样可以通过预处理,先将这些r型副产物转化为(r)-1-(3-甲氧基苯基)乙胺(即,中间体3),再按上述步骤进行回收,同样可以实现外消旋1-(3-甲氧基苯基)乙胺的回收。
[0083]
其中,后处理具体可以是:向加热回流反应结束并冷却的反应液体系中,加入酸性溶液搅拌5min,再蒸发除去低沸点溶剂,并使用有机溶剂萃取得到间甲氧基苯乙酮,再向水相加入碱性溶液调节ph至碱性,使用有机溶剂萃取得到外消旋1-(3-甲氧基苯基)乙胺,从而实现外消旋1-(3-甲氧基苯基)乙胺的回收(其中,酸性溶液可选自:硫酸溶液、盐酸溶液;碱性溶液可选自:氢氧化钠溶液、氢氧化钾溶液、碳酸钠溶液)。
[0084]
以下为具体实施例:
[0085]
实施例3(r)-1-(3-甲氧基苯基)乙胺的外消旋化反应
[0086]
合成路线如下:
[0087][0088]
本实施例所采用的具体步骤如下:
[0089]
将中间体2溶于氢氧化钠溶液(2m,200ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用50g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(r)-1-(3-甲氧基苯基)乙胺(中间体3,9.2g,61mmol)。
[0090]
在室温下,将500ml两颈烧瓶连接分水器和回流冷凝管后使用油泵抽真空并使用氮气球置换氮气氛围,依次加入对二甲苯(120ml)、中间体3(9.2g,61mmol)、间甲氧基苯乙酮(13.5g,90mmol)、钛酸四异丙酯(8.5g,30mmol),100℃磁力搅拌30h,冷却至室温后,旋转蒸发除去甲苯并转移至200ml封管中,再依次加入叔丁醇(90ml)、二甲亚砜(19.1g,240mmol)、叔丁醇钾(6.7g,60mmol),140℃搅拌8h,冷却至室温后,加入硫酸溶液(1m)调节反应液ph至2左右并搅拌5min,旋转蒸发浓缩反应液,用砂芯漏斗抽滤除去不溶物后,用乙酸乙酯洗涤三次(3
×
50ml),水相用氢氧化钠溶液(2m)调节至ph≈10,用乙酸乙酯萃取三次(3
×
25ml),合并有机相,使用30g无水硫酸钠干燥后旋转蒸发浓缩得到1-(3-甲氧基苯基)乙胺。
[0091]
外消旋回收总收率为72%。
[0092]
1-(3-甲氧基苯基)乙胺衍生的(1-(3-甲氧基苯基)乙基)氨基甲酸叔丁酯,其ee值是通过高效液相色谱仪检测的,测试条件:as-h手性柱、流动相为正己烷:异丙醇(99.2:0.8)、流速0.8ml/min、柱温25℃。hplc谱图如图2所示,其中r型出峰在19分钟左右,s型出峰在21分钟左右。峰面积表明产物的ee值小于5。
[0093]
实施例4(r)-1-(3-甲氧基苯基)乙胺的外消旋化反应
[0094]
合成路线如下:
[0095][0096]
本实施例所采用的具体步骤如下:
[0097]
将中间体2溶于氢氧化钠溶液(2m,200ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用50g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(r)-1-(3-甲氧基苯基)乙胺(中间体3,9.2g,61mmol)。
[0098]
在室温下,将250ml两颈烧瓶连接分水器和回流冷凝管后使用油泵抽真空并使用氮气球置换氮气氛围,依次加入甲苯(40ml)、中间体3(9.2g,61mmol)、间甲氧基苯乙酮(4.5g,30mmol)、钛酸四异丙酯(34.7g,122mmol),90℃磁力搅拌48h,冷却至室温后,旋转蒸发除去甲苯,再将其加入封管中,并依次加入乙二醇(180ml)、二甲亚砜(23.4g,300mmol)、叔丁醇钾(33.7g,300mmol),180℃回流搅拌6h,冷却至室温后,加入硫酸溶液(1m)调节反应液ph至2左右并搅拌5min,旋转蒸发浓缩反应液,用砂芯漏斗抽滤除去不溶物后,用乙酸乙酯洗涤三次(3
×
50ml),水相用氢氧化钠溶液(2m)调节至ph≈10,用乙酸乙酯萃取三次(3
×
25ml),合并有机相,使用30g无水硫酸钠干燥后旋转蒸发浓缩得到1-(3-甲氧基苯基)乙胺。
[0099]
外消旋回收总收率为77%。
[0100]
hplc谱图如图3所示,其中r型出峰在19分钟左右,s型出峰在22分钟左右。峰面积表明产物的ee值小于10。
[0101]
实施例5(r)-1-(3-甲氧基苯基)乙胺的外消旋化反应
[0102]
合成路线如下:
[0103][0104]
本实施例所采用的具体步骤如下:
[0105]
将中间体2溶于氢氧化钠溶液(2m,200ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用50g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(r)-1-(3-甲氧基苯基)乙胺(中间体3,9.2g,61mmol)。
[0106]
在室温下,将250ml两颈烧瓶连接分水器和回流冷凝管后使用油泵抽真空并使用氮气球置换氮气氛围,依次加入氯苯(70ml)、中间体3(9.2g,61mmol)、间甲氧基苯乙酮(15g,100mmol)、钛酸四异丙酯(51.2g,180mmol),130℃磁力搅拌30h,冷却至室温后,旋转蒸发除去甲苯,再依次加入叔戊醇(150ml)、二甲亚砜(15.6g,200mmol)、叔丁醇钾(22.4g,200mmol),80℃回流搅拌24h,冷却至室温后,加入硫酸溶液(1m)调节反应液ph至2左右并搅拌5min,旋转蒸发浓缩反应液,用砂芯漏斗抽滤除去不溶物后,用乙酸乙酯洗涤三次(3
×
50ml),水相用氢氧化钠溶液(2m)调节至ph≈10,用乙酸乙酯萃取三次(3
×
25ml),合并有机相,使用30g无水硫酸钠干燥后旋转蒸发浓缩得到1-(3-甲氧基苯基)乙胺。
[0107]
外消旋回收总收率为85%。
[0108]
hplc谱图如图4所示,其中r型出峰在19分钟左右,s型出峰在22分钟左右。峰面积表明产物的ee值小于8。
[0109]
实施例6(r)-1-(3-甲氧基苯基)乙胺的外消旋化反应
[0110]
合成路线如下:
[0111][0112]
本实施例所采用的具体步骤如下:
[0113]
将中间体2溶于氢氧化钠溶液(2m,200ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用50g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(r)-1-(3-甲氧基苯基)乙胺(中间体3,9.2g,61mmol)。
[0114]
在室温下,将250ml两颈烧瓶连接分水器和回流冷凝管后使用油泵抽真空并使用氮气球置换氮气氛围,依次加入对二甲苯(80ml)、中间体3(9.2g,61mmol)、间甲氧基苯乙酮(15g,100mmol)、钛酸四异丙酯(45.5g,160mmol),140℃磁力搅拌30h,冷却至室温后,旋转蒸发除去甲苯,将其加入封管中,再依次加入叔戊醇(150ml)、二甲亚砜(11.7g,150mmol)、叔丁醇钾(16.8g,150mmol),150℃回流搅拌15h,冷却至室温后,加入硫酸溶液(1m)调节反应液ph至2左右并搅拌5min,旋转蒸发浓缩反应液,用砂芯漏斗抽滤除去不溶物后,用乙酸乙酯洗涤三次(3
×
50ml),水相用氢氧化钠溶液(2m)调节至ph≈10,用乙酸乙酯萃取三次(3
×
25ml),合并有机相,使用30g无水硫酸钠干燥后旋转蒸发浓缩得到1-(3-甲氧基苯基)乙胺。
[0115]
外消旋回收总收率为85%。
[0116]
hplc谱图如图5所示,其中r型出峰在18分钟左右,s型出峰在21分钟左右。峰面积表明产物的ee值小于20。
[0117]
实施例7(r)-1-(3-甲氧基苯基)乙胺的外消旋化反应
[0118]
合成路线如下:
[0119][0120]
本实施例所采用的具体步骤如下:
[0121]
将中间体2溶于氢氧化钠溶液(2m,200ml)并使用乙酸乙酯萃取3次(3
×
100ml)。合并有机相,用50g无水硫酸钠干燥1h,过滤后旋转蒸发除去溶剂得到(r)-1-(3-甲氧基苯基)乙胺(中间体3,9.2g,61mmol)。
[0122]
在室温下,将250ml两颈烧瓶连接分水器和回流冷凝管后使用油泵抽真空并使用氮气球置换氮气氛围,依次加入甲苯(120ml)、中间体3(9.2g,61mmol)、间甲氧基苯乙酮(9.1g,61mmol)、钛酸四异丙酯(17.3g,61mmol),120℃磁力搅拌17h,冷却至室温后,旋转蒸发除去甲苯,再依次加入叔戊醇(120ml)、二甲亚砜(9.5g,122mmol)、叔丁醇钾(13.7g,122mmol),100℃回流搅拌4h,冷却至室温后,加入硫酸溶液(1m)调节反应液ph至2左右并搅拌5min,旋转蒸发浓缩反应液,用砂芯漏斗抽滤除去不溶物后,用乙酸乙酯洗涤三次(3
×
50ml),水相用氢氧化钠溶液(2m)调节至ph≈10,用乙酸乙酯萃取三次(3
×
25ml),合并有机相,使用30g无水硫酸钠干燥后旋转蒸发浓缩得到1-(3-甲氧基苯基)乙胺。
[0123]
外消旋回收总收率为80%。
[0124]
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1