用于检测碱基编辑器编辑位点的方法和试剂盒
1.本技术涉及基因编辑(特别是碱基编辑)技术领域。具体而言,本技术涉及一种用于检测碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑核酸的位点的方法,以及用于实施所述方法的试剂盒。本技术还涉及用于检测碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑核酸的编辑效率或脱靶效应的方法。
背景技术:
2.2016年david liu等在crispr/cas9系统的基础上将来自大鼠的rapobec1与ncas9(d10a)蛋白相融合,研发出了胞嘧啶碱基编辑器(cytosine base editor,cbe)(komor,et al.nature 533,420-424,doi:10.1038/nature17946(2016))。其设计的编辑原理为:首先,失去部分核酸切割活性的ncas9依然能为sgrna所引导,带动与ncas9相连的rapobec1至目标靶向位点处;随后,sgrna会与目的基因的dna序列形成r环(r-loop)结构,从而使得r环中处于单链状态的非sgrna互补链dna(non-target strand)能够被apobec1所结合,将该链上一定范围内的胞嘧啶(c)脱氨成尿嘧啶(u);最后,这些尿嘧啶便可通过后续的dna复制过程完成尿嘧啶至胸腺嘧啶的转换,从而最终实现c至t(c-to-t)的碱基转换。此后,编辑效率、活性编辑窗口、可编辑序列范围等各方面得到不同程度的优化的多种新的cbe编辑系统也相继被开发出来,例如ye1-be,be4max等(kim,y.b.et al.nature biotechnology 35,371-376,doi:10.1038/nbt.3803(2017);suzuki,k.et al.nature 540,144-149,doi:10.1038/nature20565(2016))。
3.此外,2020年david liu等报导了一种rna-free的线粒体胞嘧啶碱基编辑器ddcbe(ddda-derived cbe),其实现了线粒体基因编辑的重大突破(mok,b.y.et al.nature 583,631-+,doi:10.1038/s41586-020-2477-4(2020))。此前,由于线粒体双层膜的存在,将sgrna导入线粒体仍然面临极大的挑战,严重限制了基于crispr/cas9的cbe工具在线粒体基因编辑方面的应用。相对于基于crispr/cas9的cbe工具,ddcbe的主要改变包括以下两点:一是用tale蛋白代替sgrna,实现对靶向dna链的识别,避免了sgrna难以进入线粒体的难题;二是用新发现的一种双链dna脱氨酶ddda代替apobec,将靶向位点处双链dna上的dc脱氨转变为du,最终实现dc至dt的碱基转变。
4.综上,已有多种针对细胞核或者线粒体的胞嘧啶碱基编辑系统,并且还在不断的丰富中。但其核心原理均为,在靶向的编辑位点使胞嘧啶(c)脱氨成尿嘧啶(u);最后,这些尿嘧啶便可通过后续的dna复制过程完成尿嘧啶(u)至胸腺嘧啶(t),从而最终实现c至t(c-to-t)的碱基转换。
5.自2016年david liu发展了胞嘧啶碱基编辑器(komor et al.,2016)后,2017年腺嘌呤碱基编辑器(adenine base editor,abe)(gaudelli et al.,2017)也随即问世,该技术的主要编辑原理为:cas9在sgrna的引导下到达靶向编辑位点,打开dna双链形成r-loop结构,随后与cas9融合在一起的腺嘌呤脱氨酶会将编辑窗口内的腺嘌呤脱氨形成次黄嘌呤(inosine,i)。在修复以及复制过程中,次黄嘌呤将被dna聚合酶读成g,从而最终发生腺嘌
呤(a)到鸟嘌呤(g)的转变。经过几年的发展,目前使用率较高的是abemax系统,此系统基于最初abe版本进行了突变筛选、密码子优化及引入核定位信号等一系列改进,使得靶向位点的编辑效率不断提高。2020年,david liu和jennifer a.doudna又新报道了一种具有更高活性的abe版本,并命名为abe8e(richter et al.,2020)。abe8e在abemax的基础上只保留一个tada元件,且进行了多个突变,不仅提高了酶的体外活性(lapinaite et al.,2020),且在细胞内的靶向位点的编辑效率也得到了很大的提升。
6.同样,类似于cbe编辑系统,目前开发出了多种abe编辑系统,其核心原理均为,在靶向编辑位点使腺嘌呤脱氨成次黄嘌呤;之后,这些次黄嘌呤便可通过后续的dna复制过程完成次黄嘌呤至鸟嘌呤,从而最终实现腺嘌呤(a)到鸟嘌呤(g)(a-to-g)的碱基转换。
7.此外,在2020年相继有四个课题组发展了腺嘌呤与胞嘧啶双碱基编辑系统(acbe)(grunewald et al.,2020;li et al.,2020;sakata et al.,2020;zhang et al.,2020),基本原理是将此前发展的abe和cbe技术联合,实现对同一个靶向编辑窗口内部的腺嘌呤和胞嘧啶进行同时编辑。
8.理想的基因编辑工具按设计应该只会对目的靶向位点进行编辑,但实际上不论是zfn/talen还是crispr/cas系统一直以来都被发现具有脱靶风险。所谓脱靶,即是所使用的基因编辑工具在非靶标位置进行了不必要的编辑。脱靶事件一经发生,便可能会破坏该处的基因序列或染色体结构,扰乱基因组稳定性和细胞正常功能,进而可能引发各种严重的副作用,甚至诱发癌症。故而,脱靶效应对于那些对基因编辑效果的安全性要求较高的应用(比如临床治疗相关的应用)而言是基因编辑技术的一大致命缺点。如若需将碱基编辑器应用于实际,其脱靶效应必须事先进行彻底、全面且准确的检测评估。
9.理论上要检测碱基编辑器的脱靶效应,最简单直接的办法就是通过全基因组测序(whole genome sequencing,wgs)直接检测出由碱基编辑器产生的单核苷酸突变。但众所周知wgs具有很多自身方法限制:一是基因组中天然存在很多的单核苷酸变异(single nucleotide variations,snvs),dna复制过程以及后期高通量测序过程也会产生不少的随机误差,这些都会造成影响检测准确性的基因组背景(genomic background),使得wgs在检测单核苷酸突变方面灵敏度极低;二是使用高通量测序技术对全基因组进行wgs测序时,其测序读段(reads)的覆盖度(coverage)非常不均一,往往需要耗费极大的数据量才能获取足够的信息对全基因组进行评估。因此,常规的wgs并不能在全基因组水平上有效检测碱基编辑器的脱靶效应。
10.另一种方法就是,先通过软件预测(如cas-offinder等)寻找可能的脱靶位点,或者从guide-seq对crispr/cas9核酸酶系统的鉴定结果中挑选碱基编辑工具可能会造成脱靶编辑的位点,再通过定点深度测序(targeted deep sequencing)得到这些位点的准确编辑频率。所谓guide-seq,是一种通过跟踪核酸酶系统编辑过程中产生的双链断裂(double-stranded breaks,dsb)来对其脱靶位点进行检测的技术,此技术不适用于几乎不产生dsb的基因编辑技术(比如各类碱基编辑器)。通过先预测位置再进行单点深度检测的方法虽然可以从一定程度上快速获知和比较不同碱基编辑工具的脱靶风险,但其结果并不是基于全基因组水平的综合考量,得到的结论很可能因挑选的位点不同而大不相同。
11.目前用于全面评估碱基编辑系统的脱靶效应的主流技术主要有2种:一是基于体外孵育的检测技术,如digenome-seq;二是基于检测snp的技术,如goti。
12.2017年,来自韩国的jin-soo kim团队在其实验室现有的digenome-seq技术基础上针对cbe系统做了些许修改,实现了对该系统全基因组水平脱靶效应的体外检测(kim,d.et al.nature biotechnology 35,475-480,doi:10.1038/nbt.3852(2017))。其检测原理在于:首先,使用udg酶对经过be3δugi(去除ugi部分的be3)孵育的基因组dna进行处理,以期在du所在的位置产生单链断口(针对cbe),或者,使用识别di的内切酶endo v切割编辑链产生切口(针对abe),使其与由ncas9切割形成的单链断口一起形成dsb;然后,通过捕捉后续高通量测序结果中特征性的读段(reads)来获取编辑位点信息。
13.2019年杨辉团队报道了一种名为goti(genome-wide off-target analysis by two-cell embryo injection)的脱靶检测技术(zuo,e.et al.science364,289-292,doi:10.1126/science.aav9973(2019))。其技术核心在于采用了二细胞胚胎注射法,即在小鼠胚胎二细胞时期,将带有红色荧光信号的基因编辑系统注射入其中一个细胞,待胚胎发育出足够的细胞数量之后,再将整个胚胎消化成多个单细胞,并用流式细胞分选技术分别筛选出被编辑过和没被编辑过的细胞后代。理论上,红色荧光阳性细胞和阴性细胞均来自于同一枚受精卵,故而应具有相同的基因组背景,后续通过全基因组测序(wgs)对此两组细胞进行比较即可获得基因编辑造成的差异,从而获知脱靶信息。
14.就目前已有的全基因组检测技术而言,digenome-seq是一种体外检测技术,而脱靶编辑行为理论上一定会受到活细胞内真实染色质状态及局部蛋白浓度的影响,故而此技术并不能有效地反映体内环境下的真实脱靶情况。另一方面,goti等技术虽然采用了二细胞胚胎注射策略来尽量消除snv等基因组背景的影响,但也依然无法避免单细胞扩增带来的dna复制误差背景,而且此方法涉及胚胎操作,普适性不广且技术难度高、耗时长。此外,该方法依然是依赖于全基因组测序分析,要对实验涉及的所有胚胎样品均达到足够的数据覆盖率必然需要花费高额的测序费用,不适用于高通量层级的筛选评估。更重要的是,此两者方法对于碱基编辑工具的dna脱靶效应的相关结论是几乎完全相悖的,例如,kim团队发现cbe特异性很高,只会造成数量有限的cas依赖型脱靶,而杨辉团队则只鉴定到了大量的非cas依赖型脱靶。众所周知,对于脱靶效应的理解很大程度上决定了后续优化碱基编辑器的方向。对本领域而言,显然需要有一个更好、全面而没有检测偏好性的脱靶检测技术。
15.因此,亟需开发一种灵敏、无偏好性且经济适用的新型检测技术,用于在全基因组水平对碱基编辑系统的脱靶效应进行综合评估。
技术实现要素:
16.本技术的发明人基于深入的研究,开发了一种新的能够检测碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑核酸的位点、编辑效率或脱靶效应的方法。本技术的方法能够捕捉各种碱基编辑器(例如单碱基编辑器或双碱基编辑器)在编辑过程中在活细胞内产生的碱基编辑中间体,并对编辑位点进行有效标记和富集,因此,本技术的方法可普遍适用于各种碱基编辑工具的编辑位点的检测,能够评价其编辑效率或脱靶情况,且能在全基因组水平实现高灵敏度的检测。
17.因此,在一方面,本技术提供了一种检测碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑靶核酸的编辑位点、编辑效率或脱靶效应的方法,其包含下述步骤:
18.(1)提供碱基编辑器编辑靶核酸的编辑产物,其包含碱基编辑中间体,所述碱基编
辑中间体包含第一核酸链和第二核酸链;其中,所述第一核酸链包含因所述碱基编辑器编辑靶核酸而生成的编辑碱基;
19.(2)在所述第一核酸链中,在包含所述编辑碱基的区段内(例如,在所述编辑碱基的上游10nt至下游10nt的区段内)产生单链断裂切口;
20.(3)在所述单链断裂切口处或其下游引入经第一标记分子标记的核苷酸,产生含有第一标记分子的标记产物;
21.(4)分离或富集所述标记产物;例如,使用能够特异性识别和结合所述第一标记分子的第一结合分子来分离或富集所述标记产物;
22.(5)测定所述标记产物的序列;
23.从而,确定所述碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。
24.本技术的方法可以用于检测各种碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。在某些优选的实施方案中,所述碱基编辑器为单碱基编辑器或双碱基编辑器。在某些优选的实施方案中,所述碱基编辑器选自胞嘧啶单碱基编辑器,腺嘌呤单碱基编辑器,以及腺嘌呤与胞嘧啶双碱基编辑器。
25.本技术的方法不受所编辑的靶核酸限制。在某些优选的实施方案中,所述靶核酸为基因组核酸。在某些优选的实施方案中,所述靶核酸为线粒体核酸。
26.在某些优选的实施方案中,步骤(1)所述的编辑产物是所述碱基编辑器在细胞外、在细胞内或者在细胞器(例如细胞核或线粒体)内编辑靶核酸的产物。
27.在某些优选的实施方案中,所述方法在步骤(1)之前还包括如下步骤:在允许所述碱基编辑器编辑靶核酸的条件下,将所述碱基编辑器与所述靶核酸接触,从而生成所述编辑产物。所述允许碱基编辑器编辑靶核酸的条件可以是任何适宜所用碱基编辑器发挥其编辑活性的条件。
28.在某些优选的实施方案中,在允许所述碱基编辑器编辑靶核酸的条件下,在细胞外、在细胞内或者在细胞器(例如细胞核或线粒体)内,将所述碱基编辑器与所述靶核酸接触,从而生成所述编辑产物。
29.例如,所述方法在步骤(1)之前还包括如下步骤:将所述碱基编辑器导入细胞内或者细胞器内,使得所述碱基编辑器与细胞内或者细胞器内的靶核酸接触并进行碱基编辑,从而生成编辑产物;或者,将编码所述碱基编辑器的核酸分子导入细胞内或者细胞器内并使其表达所述碱基编辑器,所述碱基编辑器与细胞内或者细胞器内的靶核酸接触并进行碱基编辑,从而生成编辑产物。
30.在某些优选的实施方案中,在步骤(1)中,从所述细胞内或者细胞器内提取或分离经碱基编辑的靶核酸,并任选地,进行片段化,从而获得所述编辑产物。
31.所述片段化可采用任何适于核酸片段化的方式进行,例如通过超声或随机酶解的方法。在某些实施方案中,在进行片段化的情况下,所述编辑产物可以是含有或者不含有悬突末端的核酸片段。在某些优选的实施方案中,所述片段化(例如使用核酸内切酶的片段化)产生含有悬突末端(例如粘性末端)的核酸片段。在此类实施方案中,任选地,对含有悬突末端的核酸片段进行末端修复,生成具有平末端的核酸片段,其可用作编辑产物用于下一步骤。例如,所述末端修复可包括5’末端悬突的补平(例如通过核酸聚合反应)和/或3’末端悬突的切除。在某些优选的实施方案中,所述末端修复包括5’末端悬突的补平(例如通过
核酸聚合反应)。
32.在某些优选的实施方案中,所述第二核酸链未发生碱基编辑或不含有编辑碱基。
33.然而,易于理解的是,由于脱靶情况的存在,碱基编辑器可能在多个编辑位点(包括靶向编辑位点和脱靶位点)发生碱基编辑。例如,碱基编辑器可能对基因组dna或细胞器dna(例如,线粒体dna)的两条核酸链都进行编辑。因此,在某些情况下,所述第二核酸链潜在可能发生了碱基编辑,可能含有编辑碱基。因此,在某些实施方案中,所述第二核酸链发生了碱基编辑和/或含有编辑碱基。
34.在某些优选的实施方案中,所述编辑碱基选自尿嘧啶或次黄嘌呤。
35.在某些优选的实施方案中,步骤(2)中,在所述编辑碱基的位置处或其上游(例如上游10nt内,9nt内,8nt内,7nt内,6nt内,5nt内,4nt内,3nt内,2nt内,1nt内)或下游(例如,下游10nt内,9nt内,8nt内,7nt内,6nt内,5nt内,4nt内,3nt内,2nt内,1nt内)产生单链断裂切口。
36.在某些优选的实施方案中,在进行步骤(2)之前,所述方法还包括:修复所述编辑产物中可能存在的单链断裂(ssb)(例如内源性单链断裂)的步骤。例如,在进行步骤(2)之前,所述方法还包括:使用核酸聚合酶、核苷酸(例如不含有标记的核苷酸;例如不含有标记的dntp)和核酸连接酶(例如dna连接酶)来修复所述编辑产物中可能存在的ssb(例如内源性ssb)。
37.例如,在进行步骤(2)之前,所述方法还包括:(i)在允许核酸聚合的条件下,将所述编辑产物与核酸聚合酶(例如dna聚合酶)和核苷酸分子(优选地,不含有标记的dntp)孵育;和,(ii)使用核酸连接酶(例如dna连接酶)连接步骤(i)的产物中的缺口。在某些优选的实施方案中,所述核酸聚合酶(例如dna聚合酶)具有链置换活性。
38.不受理论限制,在步骤(2)之前进行ssb的修复是有利的。例如,ssb的修复可以消除所述编辑产物中可能存在的缺口,包括,内源存在的ssb,以及,核酸操作(例如核酸片段化)可能引入的ssb。由此,可以避免在后续步骤中在这些预先存在的ssb处或其下游引入经第一标记分子标记的核苷酸,避免这些预先存在的ssb对检测结果的干扰。
39.在某些优选的实施方案中,在步骤(2)中,使用核酸内切酶(例如,核酸内切酶v,核酸内切酶viii或ap核酸内切酶)在所述第一核酸链中产生单链断裂切口。
40.在某些优选的实施方案中,所述经第一标记分子标记的核苷酸选自,经第一标记分子标记的尿嘧啶脱氧核糖核苷酸(例如经第一标记分子标记的dutp),经第一标记分子标记的胞嘧啶脱氧核糖核苷酸(例如经第一标记分子标记的dctp),经第一标记分子标记的胸腺嘧啶脱氧核糖核苷酸(例如经第一标记分子标记的dttp),经第一标记分子标记的腺嘌呤脱氧核糖核苷酸(例如经第一标记分子标记的datp),经第一标记分子标记的鸟嘌呤脱氧核糖核苷酸(例如经第一标记分子标记的dgtp),或其任何组合。
41.在某些优选的实施方案中,所述经第一标记分子标记的核苷酸为经第一标记分子标记的尿嘧啶脱氧核糖核苷酸(例如经第一标记分子标记的dutp)或经第一标记分子标记的鸟嘌呤脱氧核糖核苷酸(例如经第一标记分子标记的dgtp)。
42.在某些优选的实施方案中,所述第一标记分子与所述第一结合分子构成了能够发生特异性相互作用(例如,能够特异性相互结合)的分子对。此类能够发生特异性相互作用(例如,能够特异性相互结合)的分子对是本领域技术人员熟知的,例如,生物素或其功能性
变体-亲和素或其功能性变体(例如生物素-亲和素,生物素-链霉亲和素),抗原/半抗原-抗体,酶和辅因子,受体-配体,能够发生点击化学反应的分子对(例如含炔基基团-叠氮基化合物)等。在某些优选的实施方案中,所述第一标记分子为生物素或其功能性变体,且所述第一结合分子为亲和素或其功能性变体;或者,所述第一标记分子为半抗原或抗原,且所述第一结合分子为特异性抗所述半抗原或抗原的抗体;或者,所述第一标记分子为含炔基基团(例如乙炔基),且所述第一结合分子为能与所述炔基(例如乙炔基)发生点击化学反应的叠氮基化合物。例如,所述经第一标记分子标记的核苷酸为含有乙炔基的核苷酸(例如,5-ethynyl-dutp),且所述第一结合分子为能与所述乙炔基发生点击化学反应的叠氮基化合物(例如叠氮基修饰的磁珠(azide magenetic beads))。
43.在某些优选的实施方案中,所述经第一标记分子标记的核苷酸中,所述第一标记分子与核苷酸的连接为可逆的或不可逆的。
44.在某些优选的实施方案中,所述经第一标记分子标记的核苷酸中,所述第一标记分子与核苷酸的连接为可逆的。在此类实施方案中,在进行步骤(4)之后,所述方法还可以包括,从所述标记产物中去除第一标记分子的步骤。在某些情况下,第一标记分子的去除是有利的,例如,可以避免对后续的扩增和/或测序步骤的不利影响。
45.在某些优选的实施方案中,所述经第一标记分子标记的核苷酸中,所述第一标记分子与核苷酸的连接为不可逆的。在此类实施方案中,优选地,所述第一标记分子的存在不会不利地影响标记产物的扩增和/或测序。例如,在某些优选的实施方案中,步骤(3)中产生的标记产物能够进行核酸扩增反应。例如,所述标记产物能够在核酸聚合酶(例如高保真或低保真核酸聚合酶)的作用下进行核酸扩增反应。
46.在某些优选的实施方案中,通过核酸聚合反应将所述经第一标记分子标记的核苷酸引入所述单链断裂切口处或其下游,从而产生含有第一标记分子的标记产物。例如,在步骤(3)中,使用核酸聚合酶(例如,具有链置换活性的核酸聚合酶)将所述经第一标记分子标记的核苷酸引入所述单链断裂切口处或其下游。例如,在步骤(3)中,在允许核酸聚合的条件下,将所述第一核酸链与核酸聚合酶和所述经第一标记分子标记的核苷酸孵育;其中,所述核酸聚合酶在所述单链断裂切口处以第二核酸链为模板起始延伸反应,并将所述经第一标记分子标记的核苷酸掺入所述单链断裂切口处或其下游。
47.在某些优选的实施方案中,步骤(3)中,所述方法还包括使用核酸连接酶(例如dna连接酶)连接所述含有第一标记分子的标记产物中缺口的步骤。
48.在某些优选的实施方案中,在步骤(3)中,在所述单链断裂切口处或其下游还引入经第二标记分子标记的核苷酸,从而产生含有第一标记分子和第二标记分子的标记产物。
49.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸是这样的核苷酸分子,其在不同的条件下(例如,经历处理前后)能够与不同的核苷酸进行碱基互补配对。例如,所述经第二标记分子标记的核苷酸在经历处理前能够与第一核苷酸进行碱基互补配对,且在经历处理后能够与第二核苷酸进行碱基互补配对。
50.在某些优选的实施方案中,所述含有第二标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸),和dac4c(n4-乙酰基胞嘧啶脱氧核糖核苷酸)。
51.在某些优选的实施方案中,所述含有第二标记的核苷酸分子为经修饰的胞嘧啶脱
氧核糖核苷酸,其在经历处理前能够与第一核苷酸(例如鸟嘌呤脱氧核糖核苷酸)进行碱基互补配对,且在经历处理后能够与第二核苷酸(例如腺嘌呤脱氧核糖核苷酸)进行碱基互补配对。在某些优选的实施方案中,所述含有第二标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸)和dac4c(n4-乙酰基胞嘧啶脱氧核糖核苷酸)。
52.例如,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。5-醛基胞嘧啶脱氧核糖核苷酸在用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)处理之前能够与鸟嘌呤脱氧核糖核苷酸进行碱基互补配对,而在用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)处理之后能够与腺嘌呤脱氧核糖核苷酸进行碱基互补配对(参见例如,liu,y.et al.bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution.nature biotechnology 37,424-429,doi:10.1038/s41587-019-0041-2(2019).;专利文献wo2015043493a1,所述参考文献全文通过引用并入本文)。
53.例如,所述经第二标记分子标记的核苷酸为5-羧基胞嘧啶脱氧核糖核苷酸。5-羧基胞嘧啶脱氧核糖核苷酸在用化合物(例如硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷))处理之前能够与鸟嘌呤脱氧核糖核苷酸进行碱基互补配对,而在用化合物(例如硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷))处理之后能够与腺嘌呤脱氧核糖核苷酸进行碱基互补配对(参见例如,liu,y.et al.bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution.nature biotechnology 37,424-429,doi:10.1038/s41587-019-0041-2(2019).,其全文通过引用并入本文)。
54.例如,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。5-羟甲基胞嘧啶脱氧核糖核苷酸可在氧化剂(例如钌酸钾)或氧化酶(例如,tet(ten-eleven translocation)蛋白)的催化下变成5-醛基胞嘧啶脱氧核糖核苷酸,而5-醛基胞嘧啶脱氧核糖核苷酸在用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)处理之前能够与鸟嘌呤脱氧核糖核苷酸进行碱基互补配对,而在用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)处理之后能够与腺嘌呤脱氧核糖核苷酸进行碱基互补配对。
55.例如,所述经第二标记分子标记的核苷酸为n4-乙酰基胞嘧啶脱氧核糖核苷酸(dac4c)。n4-乙酰基胞嘧啶脱氧核糖核苷酸在用化合物(例如氰基硼氢化钠)处理之前能够与鸟嘌呤脱氧核糖核苷酸进行碱基互补配对,而在用化合物(例如氰基硼氢化钠)处理之后能够与腺嘌呤脱氧核糖核苷酸进行碱基互补配对(参见例如,nature 583,638-643(2020),doi:10.1038/s41586-020-2418-2,其全文通过引用并入本文)。
56.在某些优选的实施方案中,通过核酸聚合反应将所述经第一标记分子标记的核苷酸和所述经第二标记分子标记的核苷酸引入在所述单链断裂切口处或其下游,从而产生含有第一标记分子和第二标记分子的标记产物。例如,在步骤(3)中,在允许核酸聚合的条件下,将所述第一核酸链与核酸聚合酶(例如,具有链置换活性的核酸聚合酶)和所述经第一
标记分子标记的核苷酸以及所述经第二标记分子标记的核苷酸孵育;其中,所述核酸聚合酶在所述单链断裂切口处以第二核酸链为模板起始延伸反应,并将所述经第一标记分子标记的核苷酸和所述经第二标记分子标记的核苷酸掺入所述单链断裂切口处或其下游。在某些优选的实施方案中,步骤(3)中,所述方法还包括使用连接酶连接所述含有第一标记分子和第二标记分子的标记产物中缺口的步骤。
57.可以理解的是,所述经第一标记分子标记的核苷酸和所述引入经第二标记分子标记的核苷酸可以在同一核酸聚合反应中引入,也可以在不同的核酸聚合反应中引入,只要能产生含有第一标记分子和第二标记分子的标记产物即可。
58.在某些实施方案中,经第二标记分子标记的核苷酸的使用或掺入是有利的。易于理解,经第二标记分子标记的核苷酸可通过核酸聚合反应通过碱基互补配对的方式掺入标记产物中。在此情况下,经第二标记分子标记的核苷酸(例如5-醛基胞嘧啶脱氧核糖核苷酸)通过与第一碱基(例如鸟嘌呤脱氧核糖核苷酸)的互补配对能力而掺入标记产物中。随后,可对标记产物进行处理(例如,用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)进行处理),由此,标记产物中的经第二标记分子标记的核苷酸将被修饰或改变,并与第二碱基(例如腺嘌呤脱氧核糖核苷酸)进行碱基互补配对。因此,当对经处理的标记产物进行测序时,经第二标记分子标记的核苷酸的掺入位置处的核苷酸将与第二碱基配对,并在测序结果中被读取为第二碱基的互补碱基(而非第一碱基的互补碱基)。换言之,在经处理的标记产物的测序结果中,在掺入经第二标记分子标记的核苷酸的位置处将产生第一碱基的互补碱基至第二碱基的互补碱基的碱基突变信号(例如c-to-t的突变信号)。通过检测该碱基突变信号,即可确定经第二标记分子标记的核苷酸的掺入位置,并进而可以对其邻近的编辑碱基进行精准定位。此外,通过核酸聚合反应,可以将一个或多个经第二标记分子标记的核苷酸掺入标记产物中,由此,在经处理的标记产物的测序结果中,将检测到一个或多个碱基突变信号。这可以放大碱基突变信号,提高检测的灵敏度。
59.因此,在使用经第二标记分子标记的核苷酸的实施方案中,优选地,在步骤(3)之后,对标记产物进行处理,以改变其包含的经第二标记分子标记的核苷酸的碱基互补配对能力。
60.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为经修饰的胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,对标记产物进行处理,以改变其包含的经修饰的胞嘧啶脱氧核糖核苷酸的碱基互补配对能力(例如,使之与腺嘌呤脱氧核糖核苷酸配对,而非与鸟嘌呤脱氧核糖核苷酸配对)。
61.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)对标记产物进行处理,以改变其包含的5-醛基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
62.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羧基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,用化合物(例如硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷))对标记产物进行处理,以改变其包含的5-羧基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
63.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,所述标记产物先用氧化剂(例如钌酸钾)或氧化酶(例如,tet蛋白)进行处理,再用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)进行处理,以改变其包含的5-羟甲基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
64.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为n4-乙酰基胞嘧啶脱氧核糖核苷酸(dac4c)。在此类实施方案中,在步骤(3)之后,用化合物(例如氰基硼氢化钠)对标记产物进行处理,以改变其包含的n4-乙酰基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
65.优选地,对标记产物的处理步骤在对标记产物进行测序之前进行,例如,在步骤(4)之前或在步骤(5)之前进行。
66.在某些情况下,经第二标记分子标记的核苷酸(例如5-醛基胞嘧啶脱氧核糖核苷酸,5-羟甲基胞嘧啶脱氧核糖核苷酸)可能是细胞内天然存在的核苷酸。为了避免此类天然存在的经第二标记分子标记的核苷酸的不利影响(例如,导致假阳性信号),可以在步骤(3)之前(例如,在步骤(2)之前),对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护(例如,使用乙基羟胺保护内源性的5-醛基胞嘧啶脱氧核糖核苷酸,或者,使用β葡萄糖基转移酶(β-glucosyltransferase,βgt)催化的糖基化反应保护内源性的5-羟甲基胞嘧啶脱氧核糖核苷酸),以防止其碱基互补配对能力发生变化。
67.因此,在某些使用经第二标记分子标记的核苷酸(例如5-醛基胞嘧啶脱氧核糖核苷酸,5-羟甲基胞嘧啶脱氧核糖核苷酸)的实施方案中,在步骤(3)之前(例如,在步骤(2)之前),对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护。
68.例如,在某些实施方案中,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,优选地,在步骤(3)之前(例如,在步骤(2)之前),使用乙基羟胺保护内源性的5-醛基胞嘧啶脱氧核糖核苷酸。
69.例如,在某些实施方案中,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,优选地,在步骤(3)之前(例如,在步骤(2)之前),使用βgt催化的糖基化反应保护内源性的5-羟甲基胞嘧啶脱氧核糖核苷酸(参见,cell,18apr 2013,153(3):678-691,doi:10.1016/j.cell.2013.04.001,其全文通过引用并入本文)。
70.在某些情况下,经第二标记分子标记的核苷酸(例如5-羧基胞嘧啶脱氧核糖核苷酸,n4-乙酰基胞嘧啶脱氧核糖核苷酸)并非是细胞内天然存在的核苷酸,或者尽管是细胞内天然存在的核苷酸,但其含量极少。在这种情况下,在步骤(3)之前,无需再对编辑产物进行核苷酸保护处理。
71.因此,在某些使用经第二标记分子标记的核苷酸(例如5-羧基胞嘧啶脱氧核糖核苷酸,n4-乙酰基胞嘧啶脱氧核糖核苷酸)的实施方案中,在步骤(3)之前,未对编辑产物进行核苷酸保护处理。
72.在某些优选的实施方案中,在步骤(2)中,在所述编辑碱基的位置处产生单链断裂切口;并且,在步骤(3)中,在所述单链断裂切口处及其下游引入所述经第一标记分子标记的核苷酸和所述经第二标记分子标记的核苷酸,产生含有第一标记分子和第二标记分子的标记产物。
73.在某些优选的实施方案中,在步骤(2)中,在所述编辑碱基的下游产生单链断裂切口;并且,在步骤(3)中,在所述单链断裂切口处或其下游引入所述经第一标记分子标记的核苷酸,且任选地,引入经第二标记分子标记的核苷酸,从而产生含有第一标记分子和任选的第二标记分子的标记产物。
74.在某些优选的实施方案中,在步骤(4)中,使用连接至固体支持物的第一结合分子来分离或富集所述标记产物。可以使用各种合适的固体支持物来承载所述第一结合分子。例如,所述固体支持物可以选自磁珠,琼脂糖珠,或芯片。
75.在某些优选的实施方案中,在进行步骤(5)之前,所述方法还包括:对步骤(4)分离或富集的标记产物进行扩增;和/或,将步骤(4)分离或富集的标记产物构建成测序文库。
76.在某些优选的实施方案中,步骤(4)中,分离或富集所述标记产物中含有第一标记和/或第二标记的核酸单链。例如,在某些实施方案中,可以将所述标记产物进行解链处理(例如,碱处理),然后,使用能够特异性识别和结合所述第一标记分子的第一结合分子来分离或富集所述标记产物中含有第一标记和/或第二标记的核酸单链。在某些实施方案中,可以使用能够特异性识别和结合所述第一标记分子的第一结合分子来分离或富集所述标记产物,然后,将所述标记产物进行解链处理(例如,碱处理),从而获得所述标记产物中含有第一标记和/或第二标记的核酸单链。在某些优选的实施方案中,所述解链处理(例如,碱处理)在第一标记分子和第一结合分子保持结合的状态下进行。
77.在某些优选的实施方案中,在进行步骤(5)之前,使用核酸聚合酶(例如低保真核酸聚合酶和/或高保真核酸聚合酶)对步骤(4)分离或富集的标记产物进行扩增。例如,在某些优选的实施方案中,所述扩增步骤包括:
78.使用低保真核酸聚合酶进行至多5个(例如至多1个,至多2个,至多3个,至多4个,至多5个)循环的聚合酶链式反应;和,
79.使用高保真核酸聚合酶进行至少3个(例如至少3个,至少5个,至少10个,至少20个,至少30个,至少40个)循环的聚合酶链式反应。
80.可以理解的是,可以使用各种合适的方法,将步骤(4)分离或富集的标记产物构建成测序文库。此类构建测序文库的的方法不受到限制。例如,可根据所使用的测序方法,构建具有相应特征的测序文库。例如,可根据测序的需要,在所述标记产物的末端添加相应测序或扩增用寡核苷酸接头。在某些实施方案中,可以在所述标记产物的3’端添加da尾,其可以用于与含有dt尾的寡核苷酸接头连接。
81.在某些优选的实施方案中,在步骤(5)中,通过测序法(例如,第二代测序法或第三代测序法)、杂交法或质谱法测定所述标记产物的序列。
82.在某些优选的实施方案中,所述方法还包括,将步骤(5)测定的序列与参考序列进行比对,从而确定所述碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。
83.在某些优选的实施方案中,所述参考序列为未进行碱基编辑之前的靶核酸序列。例如,所述未进行碱基编辑之前的靶核酸序列可获自数据库,或者可通过测序方法获得。
84.胞嘧啶碱基编辑器及其评估
85.在一个优选的实施方案中,所述碱基编辑器为胞嘧啶碱基编辑器(例如核胞嘧啶碱基编辑器,细胞器胞嘧啶碱基编辑器)。在某些优选的实施方案中,所述胞嘧啶碱基编辑器为能够将胞嘧啶编辑为尿嘧啶的胞嘧啶碱基编辑器。关于胞嘧啶碱基编辑器的详细描
述,可参见例如andrew v.anzalone,et al.nature biotechnology 38(7),824-844,doi:10.1038/s41587-020-0561-9(2020),其全文通过引用并入本文。在某些优选的实施方案中,所述碱基编辑器为能够编辑细胞核核酸的胞嘧啶碱基编辑器或能够编辑线粒体核酸的胞嘧啶碱基编辑器。
86.在某些优选的实施方案中,所述编辑碱基为尿嘧啶。
87.在某些优选的实施方案中,所述碱基编辑中间体为含有尿嘧啶的核酸分子(例如dna分子)。
88.在某些优选的实施方案中,所述含有第二标记的核苷酸分子为经修饰的胞嘧啶脱氧核糖核苷酸,其在经历处理前能够与第一核苷酸(例如鸟嘌呤脱氧核糖核苷酸)进行碱基互补配对,且在经历处理后能够与第二核苷酸(例如腺嘌呤脱氧核糖核苷酸)进行碱基互补配对。在某些优选的实施方案中,所述含有第二标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸)和dac4c(n4-乙酰基胞嘧啶脱氧核糖核苷酸)。
89.在某些优选的实施方案中,步骤(2)中,使用ap位点特异性核酸内切酶(例如,ap核酸内切酶),在所述第一核酸链中所述编辑碱基的位置处产生单链断裂切口;并且,在步骤(3)中,在所述单链断裂切口处及其下游引入所述经第一标记分子标记的核苷酸和所述经第二标记分子标记的核苷酸,产生含有第一标记分子和第二标记分子的标记产物。随后,可如之前所述,实施步骤(4)至步骤(5),从而,确定所述胞嘧啶碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。
90.在某些优选的实施方案中,在进行步骤(2)之前,所述方法还包括在所述第一核酸链中编辑碱基的位置处形成ap位点的步骤。
91.例如,在某些优选的实施方案中,在进行步骤(2)之前,所述方法还包括:将所述编辑产物与udg(尿嘧啶-dna糖基化酶)孵育的步骤。udg能够特异识别核酸链中的尿嘧啶核苷酸,并且能够特异切除所述核苷酸上的尿嘧啶,从而在核酸链中形成ap位点(去嘌呤/去嘧啶位点)。因此,udg与编辑产物的孵育能够将第一核酸链中的编辑碱基(尿嘧啶)转变为ap位点。
92.在某些优选的实施方案中,在进行与udg孵育的步骤之前,所述方法还包括,修复所述编辑产物中可能存在的ap位点的步骤。
93.在某些优选的实施方案中,所述ap位点修复步骤包括:
94.(a)在允许ap核酸内切酶发挥其切割活性的条件下,将ap核酸内切酶与可能存在ap位点的所述编辑产物孵育;
95.(b)在允许核酸聚合的条件下,将步骤(a)的产物与核酸聚合酶(例如dna聚合酶)和核苷酸分子(例如,不含有第一标记或第二标记的核苷酸分子;例如不含有标记的dntp)孵育;
96.(c)在允许核酸连接酶发挥其连接活性的条件下,将步骤(b)的产物与核酸连接酶(例如dna连接酶)孵育,
97.从而,修复所述编辑产物中可能存在的ap位点。
98.易于理解,步骤(a)中,ap核酸内切酶能够使得所述编辑产物在可能存在的ap位点处产生单链断裂切口。步骤(b)中,所述核酸聚合酶能够在所述单链断裂切口处以第二核酸
链为模板起始延伸反应,修复步骤(a)中产生的单链断裂切口。步骤(c)中,核酸连接酶(例如dna连接酶)能够连接步骤(b)的产物中的缺口。在某些优选的实施方案中,步骤(b)中的所述核酸聚合酶(例如dna聚合酶)具有链置换活性。
99.不受理论限制,在步骤(2)之前进行ap位点的修复是有利的。例如,ap位点的修复可以消除所述编辑产物中可能存在的ap位点。由此,可以避免在后续步骤中在这些预先存在的ap位点处或其下游引入经第一标记分子标记的核苷酸和经第二标记分子标记的核苷酸,避免这些预先存在的ap位点对检测结果的干扰。
100.在某些优选的实施方案中,在步骤(3)之后,对标记产物进行处理,以改变其包含的经第二标记分子标记的核苷酸的碱基互补配对能力。在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为经修饰的胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,对标记产物进行处理,以改变其包含的经修饰的胞嘧啶脱氧核糖核苷酸的碱基互补配对能力(例如,使之与腺嘌呤脱氧核糖核苷酸配对,而非与鸟嘌呤脱氧核糖核苷酸配对)。
101.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)对标记产物进行处理,以改变其包含的5-醛基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
102.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羧基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,用化合物(例如硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷))对标记产物进行处理,以改变其包含的5-羧基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
103.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,在步骤(3)之后,所述标记产物先用氧化剂(例如钌酸钾)或氧化酶(例如,tet蛋白)进行处理,再用化合物(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)进行处理,以改变其包含的5-羟甲基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
104.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为n4-乙酰基胞嘧啶脱氧核糖核苷酸(dac4c)。在此类实施方案中,在步骤(3)之后,用化合物(例如氰基硼氢化钠)对标记产物进行处理,以改变其包含的n4-乙酰基胞嘧啶脱氧核糖核苷酸的碱基互补配对能力。
105.优选地,对标记产物的处理步骤在对标记产物进行测序之前进行,例如,在步骤(4)之前或在步骤(5)之前进行。
106.在某些实施方案中,在步骤(3)之前(例如,在步骤(2)之前),对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护。例如,在步骤(3)之前(例如,在步骤(2)之前),可使用乙基羟胺保护内源性的5-醛基胞嘧啶脱氧核糖核苷酸,或者,使用βgt催化的糖基化反应保护内源性的5-羟甲基胞嘧啶脱氧核糖核苷酸。
107.例如,在某些使用经第二标记分子标记的核苷酸(例如5-醛基胞嘧啶脱氧核糖核苷酸,5-羟甲基胞嘧啶脱氧核糖核苷酸)的实施方案中,在步骤(3)之前(例如,在步骤(2)之前),对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护。
108.例如,在某些实施方案中,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,优选地,在步骤(3)之前(例如,在步骤(2)之前),使用乙基羟胺保护内源性的5-醛基胞嘧啶脱氧核糖核苷酸。
109.例如,在某些实施方案中,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,优选地,在步骤(3)之前(例如,在步骤(2)之前),使用βgt催化的糖基化反应保护内源性的5-羟甲基胞嘧啶脱氧核糖核苷酸。
110.在某些使用经第二标记分子标记的核苷酸(例如5-羧基胞嘧啶脱氧核糖核苷酸,n4-乙酰基胞嘧啶脱氧核糖核苷酸)的实施方案中,在步骤(3)之前,未对编辑产物进行核苷酸保护处理。
111.腺嘌呤碱基编辑器及其评估
112.在一个优选的实施方案中,所述碱基编辑器为腺嘌呤碱基编辑器。在某些优选的实施方案中,所述腺嘌呤碱基编辑器为能够将腺嘌呤编辑为次黄嘌呤的腺嘌呤碱基编辑器,例如腺嘌呤碱基编辑器abe7.10、abemax、abe8e。有关腺嘌呤碱基编辑器的详细描述可参见例如,andrew v.anzalone,et al.nature biotechnology 38(7),824-844,doi:10.1038/s41587-020-0561-9(2020),其全文通过引用并入本文。
113.在某些优选的实施方案中,所述编辑碱基为次黄嘌呤。
114.在某些优选的实施方案中,所述碱基编辑中间体为含有次黄嘌呤的核酸分子(例如dna分子)。
115.在某些优选的实施方案中,步骤(2)中,使用次黄嘌呤位点特异性核酸内切酶(例如,核酸内切酶v,或者核酸内切酶viii),在所述第一核酸链中所述编辑碱基的位置处或其下游产生单链断裂切口;并且,在步骤(3)中,在所述单链断裂切口处及其下游引入所述经第一标记分子标记的核苷酸,且任选地,引入经第二标记分子标记的核苷酸,产生含有第一标记分子和任选的第二标记分子的标记产物。随后,可如之前所述,实施步骤(4)至步骤(5),从而,确定所述腺嘌呤碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。
116.在某些优选的实施方案中,步骤(2)中,使用核酸内切酶v,在所述第一核酸链中所述编辑碱基的下游产生单链断裂切口;或者,使用核酸内切酶viii,在所述第一核酸链中所述编辑碱基的位置处产生单链断裂切口。
117.在此类实施方案中,标记产物中的次黄嘌呤在测序过程中会被读取为鸟嘌呤(g),由此,标记产物的测序结果中将产生a-to-g的碱基突变信号。通过检测该碱基突变信号,即可对编辑碱基进行精准定位。因此,在此类实施方案中,经第二标记分子标记的核苷酸的使用不是必需的。因此,在某些示例性实施方案中,在步骤(3)中,在所述单链断裂切口处或其下游未引入经第二标记分子标记的核苷酸。
118.然而,易于理解的是,可以使用经第二标记分子标记的核苷酸进一步放大碱基突变信号,提高检测的灵敏度。因此,在某些示例性实施方案中,在步骤(3)中,在所述单链断裂切口处或其下游引入经第二标记分子标记的核苷酸。
119.还易于理解的是,上文对于经第二标记分子标记的核苷酸的详细描述同样适用于此处。例如,在某些优选的实施方案中,所述含有第二标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸),和dac4c(n4-乙酰基胞嘧啶脱氧核糖核苷酸)。
120.此外,如上文所述,在使用经第二标记分子标记的核苷酸的实施方案中,优选地,在步骤(3)之后,对标记产物进行处理,以改变其包含的经第二标记分子标记的核苷酸的碱基互补配对能力;和/或,在步骤(3)之前(例如,在步骤(2)之前),对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护。关于经第二标记分子标记的核苷酸的处理和保护,可参见上文中的详细描述。
121.双碱基编辑器及其评估
122.在一个优选的实施方案中,所述碱基编辑器为双碱基编辑器。
123.在某些优选的实施方案中,所述碱基编辑器为能够将胞嘧啶编辑为尿嘧啶并且将腺嘌呤编辑为次黄嘌呤的碱基编辑器。
124.在某些优选的实施方案中,所述编辑碱基为次黄嘌呤和/或尿嘧啶。
125.在某些优选的实施方案中,所述碱基编辑中间体为含有次黄嘌呤和/或尿嘧啶的核酸分子(例如dna分子)。
126.易于理解,双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸的编辑产物中也包含与单碱基编辑器(例如胞嘧啶碱基编辑器和腺嘌呤碱基编辑器)编辑靶核酸而生成的编辑碱基相同的编辑碱基,因此,上文针对胞嘧啶碱基编辑器和腺嘌呤碱基编辑器及其评估所描述的内容同样适用于腺嘌呤与胞嘧啶双碱基编辑器。
127.在某些优选的实施方案中,使用上文针对胞嘧啶碱基编辑器描述的方案来检测双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸的编辑位点、编辑效率或脱靶效应。例如,可使用所述方案来检测双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸中胞嘧啶的编辑位点、编辑效率或脱靶效应。
128.在某些优选的实施方案中,使用上文针对腺嘌呤碱基编辑器描述的方案来检测双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸的编辑位点、编辑效率或脱靶效应。例如,可使用所述方案来检测双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸中腺嘌呤的编辑位点、编辑效率或脱靶效应。
129.在一个方面,本技术还提供了一种试剂盒,其包含能够在含有编辑碱基的区段内产生单链断裂切口的酶或酶的组合,含有经第一标记分子标记的核苷酸分子和能够特异性识别并结合第一标记分子的第一结合分子;其中,所述核酸内切酶或其组合能够特异识别所述含编辑碱基的碱基编辑中间体,且能够在所述编辑碱基的上游10nt(例如,10nt,9nt,8nt,7nt,6nt,5nt,4nt,3nt,2nt,1nt)至下游10nt(例如10nt,9nt,8nt,7nt,6nt,5nt,4nt,3nt,2nt,1nt)的区段内产生磷酸二酯键断裂切口。
130.在某些优选的实施方案中,所述能够在含有编辑碱基的区段内产生单链断裂切口的酶或酶的组合为核酸内切酶v,或核酸内切酶viii。
131.在某些优选的实施方案中,所述能够在含有编辑碱基的区段内产生单链断裂切口的酶或酶的组合为udg酶和ap核酸内切酶的组合。
132.在某些优选的实施方案中,所述试剂盒还包含经第二标记分子标记的核苷酸分子,所述经第二标记分子标记的核苷酸是这样的核苷酸分子,其在不同的条件下(例如,经历处理前后)能够与不同的核苷酸进行碱基互补配对。在某些优选的实施方案中,经第二标记分子标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸),和dac4c(n4-乙酰基胞嘧啶脱
氧核糖核苷酸)。
133.在某些优选的实施方案中,所述含有第二标记的核苷酸分子为经修饰的胞嘧啶脱氧核糖核苷酸,其在经历处理前能够与第一核苷酸(例如鸟嘌呤脱氧核糖核苷酸)进行碱基互补配对,且在经历处理后能够与第二核苷酸(例如腺嘌呤脱氧核糖核苷酸)进行碱基互补配对。在某些优选的实施方案中,所述含有第二标记的核苷酸分子选自d5fc(5-醛基胞嘧啶脱氧核糖核苷酸),d5cac(5-羧基胞嘧啶脱氧核糖核苷酸),d5hmc(5-羟甲基胞嘧啶脱氧核糖核苷酸)和dac4c(n4-乙酰基胞嘧啶脱氧核糖核苷酸)。
134.在某些优选的实施方案中,所述试剂盒还包含保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺,βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物),或其任何组合),和/或,处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,叠氮茚二酮,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),钌酸钾,tet蛋白,氰基硼氢化钠,或其任何组合)。
135.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,所述试剂盒还可以包含保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺),和/或,处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),或叠氮茚二酮)。
136.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羟甲基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,所述试剂盒还可以包含保护经第二标记分子标记的核苷酸分子的试剂(例如βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物)),和/或,处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如钌酸钾或tet蛋白,和丙二腈或硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷)或叠氮茚二酮)。
137.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为5-羧基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,所述试剂盒还可以包含,处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷))。
138.在某些优选的实施方案中,所述经第二标记分子标记的核苷酸为n4-乙酰基胞嘧啶脱氧核糖核苷酸。在此类实施方案中,所述试剂盒还可以包含,处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如氰基硼氢化钠)。
139.在某些优选的实施方案中,所述试剂盒还包含核酸聚合酶(例如含有链置换活性的核酸聚合酶),核酸连接酶(例如dna连接酶),未经标记的核苷酸分子,保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺,βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物),或其任何组合),处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,叠氮茚二酮,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),钌酸钾,tet蛋白,氰基硼氢化钠,或其任何组合),或其任何组合。
140.易于理解,所述试剂盒用于实施本技术的方法。因此,上文对于碱基编辑器(例如单碱基编辑器和双碱基编辑器)、第一标记分子、第一结合分子、经第一标记分子标记的核
苷酸分子、第二标记分子、经第二标记分子标记的核苷酸分子、核酸聚合酶、核酸连接酶、udg酶、ap核酸内切酶、核酸内切酶v或viii等的详细描述同样适用于此处。
141.在某些优选的实施方案中,所述试剂盒用于检测碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑靶核酸的编辑位点、编辑效率或脱靶效应。
142.在某些优选的实施方案中,所述试剂盒用于检测胞嘧啶碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。在某些优选的实施方案中,所述试剂盒包括,udg酶,ap核酸内切酶,经第一标记分子标记的核苷酸分子,第一结合分子和经第二标记分子标记的核苷酸分子(例如d5fc,d5cac,d5hmc或dac4c);任选地还包含,核酸聚合酶,核酸连接酶,未经标记的核苷酸分子,保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺,βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物),或其任何组合),处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,叠氮茚二酮,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),钌酸钾,tet蛋白,氰基硼氢化钠,或其任何组合),或其任何组合。
143.在某些优选的实施方案中,所述试剂盒用于检测腺嘌呤碱基编辑器编辑靶核酸的编辑位点、编辑效率或脱靶效应。在某些优选的实施方案中,所述试剂盒包括,核酸内切酶v或viii,经第一标记分子标记的核苷酸分子和第一结合分子;任选地还包含,核酸聚合酶,核酸连接酶,经第二标记分子标记的核苷酸分子(例如d5fc,d5cac,d5hmc或dac4c),未经标记的核苷酸分子,保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺,βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物),或其任何组合),处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,叠氮茚二酮,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),钌酸钾,tet蛋白,氰基硼氢化钠,或其任何组合),或其任何组合。
144.在某些优选的实施方案中,所述试剂盒用于检测双碱基编辑器(例如腺嘌呤与胞嘧啶双碱基编辑器)编辑靶核酸的编辑位点、编辑效率或脱靶效应。在某些优选的实施方案中,所述试剂盒包括,udg酶,ap核酸内切酶,核酸内切酶v或viii,经第一标记分子标记的核苷酸分子,第一结合分子和经第二标记分子标记的核苷酸分子(例如d5fc,d5cac,d5hmc或dac4c);任选地还包含,核酸聚合酶,核酸连接酶,未经标记的核苷酸分子,保护经第二标记分子标记的核苷酸分子的试剂(例如乙基羟胺,βgt催化的糖基化反应所需的试剂(例如β-葡萄糖基转移酶,葡萄糖基化合物),或其任何组合),处理经第二标记分子标记的核苷酸分子以改变其碱基互补配对能力的试剂(例如丙二腈,叠氮茚二酮,硼烷类化合物(例如吡啶硼烷类化合物,例如吡啶硼烷或2-甲基吡啶硼烷),钌酸钾,tet蛋白,氰基硼氢化钠,或其任何组合),或其任何组合。
145.术语定义
146.在本技术中,除非另有说明,否则本文中使用的科学和技术名词具有本领域技术人员所通常理解的含义。并且,本文中所用的核酸化学实验室操作步骤均为相应领域内广泛使用的常规步骤。同时,为了更好地理解本发明,下面提供相关术语的定义和解释。除非在本文别处具体限定或不同地描述,否则以下与本发明有关的术语和描述应按照下面给出的定义来理解。
147.当本文使用术语“例如”、“如”、“诸如”、“包括”、“包含”或其变体时,这些术语将不
被认为是限制性术语,而将被解释为表示“但不限于”或“不限于”。
148.除非本文另外指明或根据上下文明显矛盾,否则术语“一个”和“一种”以及“该”和类似指称物在描述本发明的上下文中(尤其在以下权利要求的上下文中)应被解释成覆盖单数和复数。
149.如本文所用,术语“碱基编辑器”是指,包含能够对核酸分子(例如dna或rna)中的碱基(例如a,t,c,g或u)进行编辑或修饰的多肽的试剂。在一些实施方案中,所述碱基编辑器为单碱基编辑器或双碱基编辑器。
150.在一些实施方案中,所述碱基编辑器为单碱基编辑器,其能够编辑核酸分子(例如dna分子)内的一种碱基;例如,其能够使核酸分子(例如dna分子)内的一种碱基脱氨。在一些实施方案中,所述单碱基编辑器能够使dna中的腺嘌呤(a)脱氨。在一些实施方案中,所述单碱基编辑器能够使dna中的胞嘧啶(c)脱氨。在一些实施方案中,所述单碱基编辑器包含腺苷脱氨酶和核酸可编程dna结合蛋白(napdnabp),例如,是包含与腺苷脱氨酶融合的核酸可编程dna结合蛋白(napdnabp)的融合蛋白。在一些实施方案中,单碱基编辑器包含胞苷脱氨酶和核酸可编程dna结合蛋白(napdnabp),例如,是包含与胞苷脱氨酶融合的napdnabp的融合蛋白。在一些实施方案中,所述核酸可编程dna结合蛋白(napdnabp)为cas9蛋白,例如只能切割核酸双链体一条链的cas9 nickase(ncas9)或者无核酸酶活性的cas9(dcas9)。
151.在一些实施方案中,单碱基编辑器包含腺苷脱氨酶和cas9蛋白,例如,是与腺苷脱氨酶融合的cas9蛋白。在一些实施方案中,单碱基编辑器包含胞苷脱氨酶和cas9蛋白,例如,是与胞苷脱氨酶融合的cas9蛋白。在一些实施方案中,单碱基编辑器包含腺苷脱氨酶和ncas9,例如,是与腺苷脱氨酶融合的ncas9。在一些实施方案中,单碱基编辑器包含胞苷脱氨酶和ncas9,例如,是与胞苷脱氨酶融合的ncas9。在一些实施方案中,单碱基编辑器包含腺苷脱氨酶和dcas9,例如,是融合于腺苷脱氨酶的dcas9。在一些实施方案中,单碱基编辑器包含胞苷脱氨酶和dcas9,例如,是融合于胞苷脱氨酶的dcas9。
152.在一些实施方案中,所述碱基编辑器为双碱基编辑器,其能够编辑核酸分子(例如dna分子)内的两种碱基;例如,其能够使核酸分子(例如dna分子)内的两种碱基脱氨。在一些实施方案中,所述双碱基编辑器能够使dna中的腺嘌呤(a)和胞嘧啶(c)脱氨。在一些优选的实施方案中,所述双碱基编辑器能够使dna中位于同一编辑窗口内的腺嘌呤(a)和胞嘧啶(c)脱氨。在一些实施方案中,所述双碱基编辑器包含腺苷脱氨酶、胞苷脱氨酶和核酸可编程dna结合蛋白(napdnabp)。在一些实施方案中,所述核酸可编程dna结合蛋白(napdnabp)为cas9蛋白,例如只能切割核酸双链体一条链的cas9 nickase(ncas9)或者无核酸酶活性的cas9(dcas9)。在一些实施方案中,所述双碱基编辑器包含腺苷脱氨酶、胞苷脱氨酶和cas9蛋白。在一些实施方案中,所述双碱基编辑器包含腺苷脱氨酶、胞苷脱氨酶和cas9nickase(ncas9)。在一些实施方案中,所述双碱基编辑器包含腺苷脱氨酶、胞苷脱氨酶和无核酸酶活性的cas9(dcas9)。在一些实施方案中,所述双碱基编辑器是包含腺苷脱氨酶、胞苷脱氨酶和napdnabp的复合物或融合蛋白。
153.易于理解,所述双碱基编辑器可包含一个或多个(例如一个或两个)核酸可编程dna结合蛋白(napdnabp)。在一些实施方案中,所述双碱基编辑器包含两个napdnabp,其分别独立地与腺苷脱氨酶和胞苷脱氨酶融合。在一些实施方案中,所述双碱基编辑器包含1个napdnabp,其同时与腺苷脱氨酶和胞苷脱氨酶融合。在一些实施方案中,所述双碱基编辑器
是两种单碱基编辑器的组合。
154.在一些实施方案中,碱基编辑器被融合到碱基切除修复的抑制剂(例如ugi结构域或disn结构域)。在一些实施方案中,所述融合蛋白包含与脱氨酶融合的ncas9和碱基切除修复抑制剂,例如ugi或disn结构域。在一些实施方案中,所述碱基切除修复抑制剂,例如ugi结构域或disn结构域,在系统中被提供,但是不融合到cas9蛋白(或dcas9,ncas9)。需要强调的是,此处所述“与
…
融合”或“融合到
…”
包括使用或不使用接头进行的蛋白(或其功能结构域)之间的融合或连接。在某些实施方案中,所述“接头”是肽接头。在某些实施方案中,所述“接头”是非肽接头。
155.在一些实施方案中,所述碱基编辑器包含的脱氨酶与核酸可编程dna结合蛋白在结构上彼此独立,即,所述碱基编辑器包含的脱氨酶与核酸可编程dna结合蛋白没有通过接头进行融合或连接。在某些实施方案中,所述碱基编辑器包含的脱氨酶与核酸可编程dna结合蛋白之间非共价地连接或结合。
156.易于理解,所述脱氨酶可以是任意碱基形成的糖苷的特异性脱氨酶或其组合(例如,腺苷脱氨酶,胞苷脱氨酶)。
157.在某些实施方案中,所述核酸可编程dna结合蛋白可选自tales,zfs,casx,casy,cpf1,c2c1,c2c2,c2c3,argonaute蛋白,或其衍生形式。在某些实施方案中,所述可编程dna结合蛋白不具有核酸酶活性。在某些实施方案中,所述可编程dna结合蛋白只能切割核酸双链体中的一条链。在某些实施方案中,所述可编程dna结合蛋白不具有形成核酸双链断裂切口的活性。
158.在某些实施方案中,所述碱基编辑器是胞嘧啶碱基编辑器,例如胞嘧啶碱基编辑器be3,胞嘧啶碱基编辑器升级版be4max,线粒体胞嘧啶碱基编辑器ddcbe,以及各种cbe编辑系统。关于各种胞嘧啶碱基编辑器的描述,可参见例如,andrew v.anzalone,et al.nature biotechnology 38(7),824-844,doi:10.1038/s41587-020-0561-9(2020),其全文通过引用并入本文。
159.在某些实施方案中,所述碱基编辑器是腺嘌呤碱基编辑器,例如腺嘌呤碱基编辑器abe7.10、腺嘌呤碱基编辑器abemax和腺嘌呤碱基编辑器abe8e,以及各种abe编辑系统。关于各种腺嘌呤碱基编辑器的详细描述,可参见例如,andrew v.anzalone,et al.nature biotechnology 38(7),824-844,doi:10.1038/s41587-020-0561-9(2020),其全文通过引用并入本文。
160.在某些实施方案中,所述碱基编辑器是能够编辑腺嘌呤与胞嘧啶的碱基编辑器,例如acbe。
161.如本文所用,术语“碱基编辑中间体”是指,碱基编辑器(例如单碱基编辑器或双碱基编辑器)编辑靶核酸的产物,其包含因所述碱基编辑器编辑靶核酸而生成的编辑碱基。所述靶核酸可来源于任何生物体(例如真核细胞,原核细胞,病毒和类病毒)或非生物体(例如核酸分子文库)。在某些实施方案中,所述碱基编辑中间体是碱基编辑器编辑靶核酸的直接产物。在某些实施方案中,所述碱基编辑中间体是碱基编辑器编辑靶核酸的直接产物经富集和/或核酸片段化处理得到的产物。在某些实施方案中,所述编辑碱基是经所述碱基编辑器中相应活性元件(例如胞苷脱氨酶,腺苷脱氨酶)修饰了的碱基(例如尿嘧啶,次黄嘌呤)。通常而言,修饰/编辑前后的碱基具有不同的碱基互补配对能力(即,能与不同的碱基进行
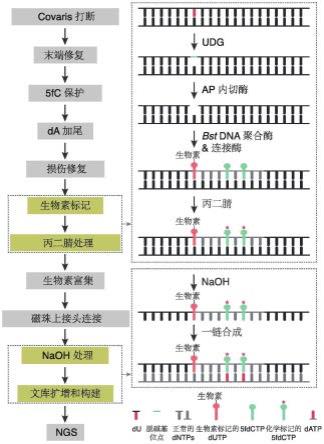
互补配对)。例如,核酸中的胞嘧啶经碱基编辑器中的胞苷脱氨酶编辑后,转变为尿嘧啶,尿嘧啶与腺嘌呤互补配对,而非鸟嘌呤。例如,核酸中的腺嘌呤经碱基编辑器中的腺苷脱氨酶编辑后,转变为次黄嘌呤,次黄嘌呤与胞嘧啶互补配对,而非胸腺嘧啶。
162.如本文所用,术语“硼烷类化合物”是指可用于对本技术的经第二标记分子标记的核苷酸进行处理,以改变其碱基互补配对能力的硼烷类化合物。特别是吡啶硼烷类化合物,其包含吡啶硼烷及其衍生物。所述吡啶硼烷类化合物的非限制性实例为吡啶硼烷、2-甲基吡啶硼烷(参见例如,liu,y.et al.bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution.nature biotechnology 37,424-429,doi:10.1038/s41587-019-0041-2(2019).,其全文通过引用并入本文)。
163.如本文中所用,术语“上游”用于描述两条核酸序列(或两个核酸分子)的相对位置关系,并且具有本领域技术人员通常理解的含义。例如,表述“一条核酸序列位于另一条核酸序列的上游”意指,当以5'至3'方向排列时,与后者相比,前者位于更靠前的位置(即,更接近5'端的位置)。如本文中所使用的,术语“下游”具有与“上游”相反的含义。
164.如本文中所用,术语“第一标记分子”是指,能与第一结合分子特异性形成相互作用分子对的分子。根据本技术的方法,第一结合分子与第一标记分子的特异性结合可用于富集所述含有第一标记分子的标记产物。在某些实施方案中,所述第一标记分子与所述第一结合分子可逆地或不可逆地结合。在某些优选的实施方式中,所述第一标记分子与所述第一结合分子可逆地结合。
165.如本文中所用,术语“经第一标记分子标记的核苷酸”是指,含有所述第一标记分子中能够与第一结合分子特异性形成相互作用分子对的基团的核苷酸分子。在一些优选的实施方案中,所述经第一标记分子标记的核苷酸是指单核苷酸分子,例如经第一标记分子标记的dutp,datp,dttp,dctp或dgtp,或其任何组合。
166.在一些实施方案中,被标记的核苷酸分子与第一标记分子之间可逆或不可逆地连接。在一些实施方案中,被标记的核苷酸分子的核糖,碱基,或磷酸部分与第一标记分子之间可逆或不可逆地连接。在一些优选的实施方案中,被标记的核苷酸分子与第一标记分子之间可逆地连接。需要留意的是,在某些情况下,经第一标记分子标记的核苷酸分子不含有第一标记分子的完整结构,但含有所述第一标记分子中能够与第一结合分子特异性形成相互作用分子对的基团。
167.如本文中所用,术语“第二标记分子”是指,能修饰核苷酸分子中的碱基以产生修饰碱基的分子,所述修饰碱基在不同的条件下(例如,经历处理前后)能够与不同的碱基互补配对。
168.如本文中所用,术语“经第二标记分子标记的核苷酸”是指,在不同的条件下(例如,经历处理前后)能够与不同的核苷酸进行碱基互补配对的核苷酸分子。在一些优选的实施方案中,所述经第二标记分子标记的核苷酸是指单核苷酸分子。
169.如本文所用,具有“链置换活性”的核酸聚合酶是指,在延伸新核酸链的过程中,如果遇到下游与模板链互补的核酸链,可以继续延伸反应并将所述与模板链互补的核酸链降解(而非剥离)的核酸聚合酶。在某些优选的实施方案中,所述具有“链置换活性”的核酸聚合酶还具有5’端至3’端外切酶活性。
170.如本文所使用的,“高保真核酸聚合酶”是指,在扩增核酸的过程中,引入错误核苷酸的概率(即,错误率)低于野生型taq酶(例如其序列如uniprot acession:p19821.1所示的taq酶)的核酸聚合酶。例如,start high-fidelity dna polymerase。
171.如本文所使用的,“低保真核酸聚合酶”是指,在扩增核酸的过程中,引入错误核苷酸的概率(即,错误率)高于野生型taq酶(例如其序列如uniprot acession:p19821.1所示的taq酶)的核酸聚合酶。例如,mightyamp dna polymerase。
172.如本文所使用的,除非上下文明确指出,否则,本文所使用的术语“核苷酸”优选是指核苷三磷酸,例如脱氧核糖核苷三磷酸。
173.有益效果
174.本技术提供了一种新的检测碱基编辑器(例如胞嘧啶碱基编辑器,腺嘌呤碱基编辑器,腺嘌呤与胞嘧啶双碱基编辑器)编辑核酸的位点、效率或脱靶效应的方法,其具有一个或多个选自下列的有益技术效果:
175.(1)本发明的方法能够捕获碱基编辑工具在活细胞内产生的碱基编辑中间体(例如含有尿嘧啶或次黄嘌呤的核酸),因此,其能够获取真实发生了碱基编辑事件的位点信息。
176.(2)本发明的方法能够对编辑位点进行有效标记和富集,从而能够非常容易地与snv、测序误差等基因背景进行区分。
177.(3)现有技术中利用全基因组测序技术对碱基编辑位点进行检测时,测序读段对全基因组的覆盖度(coverage)非常不均一,从而需要耗费极大的数据量才能获取足够的信息对全基因组中的编辑位点进行评估。本发明的方法克服了这一困难,能够在较低数据量下获取全基因组水平的强检测信号。
178.(4)本发明的方法对各种碱基编辑工具(例如cbe,abe)没有偏好性。如前所述,为满足实际需要,目前已开发出各种优化的碱基编辑工具。由于本发明的方法能够捕获各种碱基编辑过程都会产生的碱基编辑中间体(例如含有尿嘧啶或次黄嘌呤的核酸),因此,本发明的方法可普遍适用于各种碱基编辑工具的编辑位点的检测,能够评价其编辑效率或脱靶情况。
179.下面将结合附图和实施例对本发明的实施方案进行详细描述,但是本领域技术人员将理解,下列附图和实施例仅用于说明本发明,而不是对本发明的范围的限定。根据附图和优选实施方案的下列详细描述,本发明的各种目的和有利方面对于本领域技术人员来说将变得显然。
附图说明
180.图1显示了利用本发明的方法检测碱基编辑器的编辑位点的示例性方案1,其中,所述碱基编辑器是胞嘧啶碱基编辑器。
181.第一步,提取经胞嘧啶碱基编辑器编辑的核酸(例如基因组dna或线粒体dna),其含有碱基编辑中间体(例如含有尿嘧啶的dna),所述碱基编辑中间体是胞嘧啶碱基编辑器编辑靶核酸的产物,且包含第一核酸链和第二核酸链;其中,所述第一核酸链包含因胞嘧啶碱基编辑器编辑靶核酸而生成的编辑碱基(例如尿嘧啶)。通过例如超声等方法将所述核酸打断,以形成例如约300bp的核酸片段,之后通过末端修复过程将打断后的基因组dna片段
修整成平末端。在某些示例性实施方案中,所述末端修复过程包括3’末端悬突的切除过程和5’末端悬突的补平过程。在某些优选的实施方案中,所述末端修复过程可利用含有3’至5’外切活性的核酸聚合酶进行。
182.第二步,经过体外ber(碱基切除修复途径)标记法在碱基编辑中间体中编辑碱基(例如尿嘧啶)所在的位置及其下游掺入经第一标记分子(例如生物素)标记的核苷酸(例如尿嘧啶脱氧核糖核苷酸)与经第二标记分子标记的核苷酸(例如5-醛基胞嘧啶脱氧核糖核苷酸)。在某些示例性方案中,所述ber标记法包括:使用udg(尿嘧啶-dna糖基化酶)对胞嘧啶碱基编辑器编辑靶核酸产生的编辑产物上的尿嘧啶进行特异性识别与切除,产生ap位点;用ap核酸内切酶切除脱碱基位点,产生单链缺口;利用含有链置换活性的dna聚合酶从产生的单链缺口开始沿着5’至3’方向进行dna链置换反应;用dna连接酶连接dna链置换反应产物中的单链切口。其中,所述dna链置换反应体系中,使用至少一种经第一标记分子(例如生物素)标记的核苷酸底物(例如生物素-尿嘧啶核糖核苷酸)来代替常规的核苷酸酸底物(例如胸腺嘧啶脱氧核糖核苷酸)。在某些优选的实施方案中,所述dna链置换反应体系中还包括至少一种经第二标记分子标记的核苷酸底物(例如5-醛基胞嘧啶脱氧核糖核苷酸)来代替常规的核苷酸底物(例如胞嘧啶脱氧核糖核苷酸)。经第一标记分子标记的核苷酸(例如生物素-尿嘧啶脱氧核糖核苷酸)的掺入可以使得后续能够利用第一结合分子(例如链霉亲和素)富集所述含有第一标记分子的核酸片段,其中,所述第一结合分子能与所述第一标记分子能够特异性相互作用。经第二标记分子标记的核苷酸在不同的条件下(例如,经历处理前后)能够与不同的核苷酸进行碱基互补配对。例如,所述经第二标记分子标记的核苷酸为5-醛基胞嘧啶脱氧核糖核苷酸(d5fc);其在用化合物(例如丙二腈,或叠氮茚二酮)处理之前能够与鸟嘌呤脱氧核糖核苷酸进行碱基互补配对,而在用化合物(例如丙二腈,或叠氮茚二酮)处理之后能够与腺嘌呤脱氧核糖核苷酸进行碱基互补配对,由此,含有d5fc的标记产物可通过后续化学反应在掺入d5fc的位置产生c-to-t突变信号,从而实现对编辑碱基(例如,尿嘧啶)所在位置的精准定位。
183.在某些优选的实施方案中,为避免内源性或核酸操作过程中引入的dna损伤或修饰(例如,ssb或ap位点)可能带来的假阳性信号,在进行第二步之前,所述方法还包括,对编辑产物进行核酸修复处理。在某些示例性实施方案中,所述处理包括:用ap内切酶切除ap位点,以产生单链缺口;用dna聚合酶从产生的单链缺口或者核酸链中可能存在的ssb缺口处开始沿着5’至3’方向进行dna链置换反应;用dna连接酶连接链置换反应产物中的缺口。在某些优选的实施方案中,所述dna聚合酶具有链置换活性。
184.在某些优选的实施方案中,为避免内源性的经第二标记分子标记的核苷酸(例如内源性的5-醛基胞嘧啶脱氧核糖核苷酸)的不利影响,在进行第二步之前,所述方法还包括,对编辑产物进行中可能存在的经第二标记分子标记的核苷酸进行保护。例如,在进行第二步之前,可使用乙基羟胺(etonh2)对编辑产物进行中可能存在的5-醛基胞嘧啶脱氧核糖核苷酸进行保护,以防止其后续与化合物(例如丙二腈,或叠氮茚二酮)反应,形成假阳性碱基转换信号。
185.第三步,对前一步产生的含有经第二标记分子标记的核苷酸的核酸进行处理,以改变经第二标记分子标记的核苷酸的碱基互补配对能力。在某些优选的实施方案中,所述经第二标记分子标记的核苷酸是5-醛基胞嘧啶脱氧核糖核苷酸。如上所述,经化合物(例如
丙二睛,或叠氮茚二酮)处理的5-醛基胞嘧啶脱氧核糖核苷酸在后续dna复制过程中会与腺嘌呤脱氧核糖核苷酸进行碱基互补配对,从而,在所述经处理的核酸的扩增产物的测序结果中,5-醛基胞嘧啶脱氧核糖核苷酸所在的位置处会产生c-to-t的突变信号。
186.第四步,利用偶联有第一结合分子(例如链霉亲和素)的固相支持物(例如磁珠)富集含有第一标记分子(例如生物素)的dna片段;其任选地经过扩增和/或文库构建后,可用于高通量测序。根据测序结果,可分析胞嘧啶碱基编辑器编辑靶核酸后产生的碱基编辑中间体中编辑位点的位置信息。
187.在某些优选的实施方案中,在对富集的dna片段进行扩增和/或文库构建前,还可以对固相支持物(例如磁珠)上富集的dna片段进行处理(例如碱处理),以去除含有第一标记分子(例如生物素)的核酸单链的互补链。
188.在某些示例性实施方案中,在用碱(例如naoh)处理以去除含有第一标记分子(例如生物素)的核酸单链的互补链之前,通过接头连接反应在富集的dna片段末端连上寡核苷酸接头,以便于dna片段的扩增或测序。在某些优选的实施方案中,在dna片段的3’端添加da尾,所述da尾可用于与含有dt尾的寡核苷酸接头连接。
189.图2显示了本发明实施例1的方法所用不同模式序列的示意图(a),以及,本发明实施例1的方法对不同模式序列的富集结果(b)。
190.图3显示了本发明实施例1的方法在模式序列上产生的高通量测序信号。(a)含du:dg碱基对模式序列的高通量测序结果。灰色虚线指示du:dg碱基对所在的位置,红色色块即为c-to-t突变信号;(b)基于高通量测序数据对模式序列上不同位置处的c-to-t突变比例的统计计算结果。灰色虚线指示du:da碱基对所在的位置,红色实心点指示连续性c-to-t突变信号的位置,空心点指示信号低于背景水平的c所在的位置。
191.图4显示了本发明实施例1的方法在基因组dna上产生的信号。(a)在on-target位点处产生的信号。上半部指示利用本发明的方法在hek293t细胞系中由不同编辑组分和不同处理方法获得的样品在emx1on-target位点处产生的信号,下半部指示利用本发明的方法在hek293t细胞系中由不同编辑组分和不同处理方法获得的样品在vegfa_site_2on-target位点处产生的信号。样品名称中,“in”指示input样品,“nt”指示转染了be4max与non-target sgrna的样品,“rep1”指示重复1,“rep2”指示重复2;绿色的“a”等同于指示非靶向链上的c-to-t信号;(b)在全基因组水平产生的连续性c-to-t突变信号统计。左半部统计产生的突变信号距离,右半部统计产生的突变个数;(c)在vegfa_site_2样品某一脱靶位点处的信号。红色色块指示非靶向链上的“c-to-t”突变,红色倒三角指示实际被cbe编辑的位置,黑色倒三角指示“g-to-t”snv,棕色阴影指示prbs,即推测的sgrna结合位点(putative sgrna binding site);(d)prbs(深蓝色)或随机位点(浅绿色)前后4kb范围内的本发明信号(左)与wgs信号(右)对比。
192.图5显示了对cbe系统进行不同组分删除对比实验所用质粒构成的示意图。
193.图6显示了非cas依赖型脱靶的检测结果。(a)不同样品中非cas依赖型脱靶位点的信号示例。(-)sgrna样品中的红色“t”指示着本发明的方法产生的c-to-t信号,此信号在其他样品中并未观测到;(b)在不同样品中鉴定到的非cas依赖型脱靶位点个数;(c)在各个all与(-)sgrna样品中鉴定到的非cas依赖型脱靶位点的交集情况;(d)不同样品中此类非cas依赖型脱靶位点处的序列基序分析结果。每个位点两侧10bp的邻近序列(参照hg38基因
组)均被提取并通过weblogo软件进行序列分析;(e)本发明的方法鉴定到的非cas依赖型脱靶位点富集出现在基因组转录活跃区域;(f)本发明鉴定到的非cas依赖型脱靶位点更集中出现在高表达基因区。所有p值均通过单边student’s t-test计算获得。
194.图7显示了cas依赖型脱靶的检测结果。(a)不同样品中cas依赖型脱靶位点的信号示例。右侧放大的igv(integrative genomics viewer)图中,绿色色块即为“g-to-a”突变,等同于非靶向链上的“c-to-t”突变;(b)在“vegfa_site_2-all”两个生物学重复样品中鉴定到的cas依赖型脱靶位点。在非常严格的生信分析鉴定规则(cufoff)下,判定为重复出现的位点为384个(橙色点;包括on-target在内),但实际上剩下的rep-only点(蓝色点)的信号强度在两个样品中都不低;(c)全基因组水平所有cas依赖型脱靶位点的本发明信号在不同样品中的比较。细胞内天然存在的内源du修饰(灰色点)信号基本保持在对角线位置上不变,而on-target位点(红色点)以及cas依赖型脱靶位点(橙色点)信号强度随着去除的组分而变化。
195.图8显示了本发明实施例1的方法检测的信号强度与定点深度测序结果的对比。ρ即为spearman相关系数。注:图中展示皆为cas依赖型脱靶位点的验证数据。
196.图9显示了通过定点深度测序法验证本发明方法检测到cas依赖型脱靶的两个示例。(a)不同样品中“vegfa_site_2prbs-237”脱靶位点处的真实编辑效率;(b)不同样品中“vegfa_site_2prbs-67”脱靶位点处的真实编辑效率。
197.图10显示了利用本发明的方法在全基因组水平检测到的“emx1”、“vegfa_site_2”与“hek293 site_4”sgrna靶向编辑位点和cas依赖型脱靶编辑位点在各染色体上的分布。靶向编辑位点和cas依赖型脱靶编辑位点分别由红色正方形和蓝色圆圈指示。
198.图11显示了本发明实施例1的方法与guide-seq(a)和digenome-seq(b)检测到的cas依赖型脱靶位点作比较的venn图。
199.图12显示了使用本发明方法对cbe优化工具ye1-be4max特异性的再评估检验结果。(a)全基因组水平所有cas依赖型脱靶位点的检测信号在ye1-be4max(纵轴)与wt-be4max(横轴)样品中的比较;(b)不同位点处ye1-be4max与wt-be4max的编辑效率。红色三角指示剩余大量脱靶编辑的位置。
200.图13显示了本发明实施例1的方法检测到的对于“runx1”与“dyrk1a”位点由lbcpf1-be在全基因组水平造成的cas依赖型脱靶。横纵坐标为本发明在两个生物学重复样品中鉴定到的信号强度。
201.图14显示了利用本发明实施例1的方法检测到的crispr-free的ddcbe工具造成的tale依赖型脱靶(a)和非tale依赖型脱靶(b)示例。上图为放大的igv(integrative genomics viewer)图,红色色块为“c-to-t”突变,绿色色块即为“g-to-a”突变,等同于互补链上的“c-to-t”突变;中图mcherry为阴性对照样品;下图为通过定点深度测序法验证本发明方法检测到脱靶位点的测序结果。
202.图15显示了利用本发明的方法检测碱基编辑器的编辑位点的示例性方案2,其中,所述碱基编辑器是腺嘌呤碱基编辑器。
203.首先,第一步提取经腺嘌呤碱基编辑器编辑的核酸(例如基因组dna),其含有碱基编辑中间体(例如含有次黄嘌呤的dna),所述碱基编辑中间体是腺嘌呤碱基编辑器编辑靶核酸的产物,且包含第一核酸链和第二核酸链;其中,所述第一核酸链包含因腺嘌呤碱基编
辑器编辑靶核酸而生成的编辑碱基(例如次黄嘌呤)。通过例如超声等方法将所述核酸打断,以形成例如约300bp的核酸片段,之后通过末端修复过程将打断后的基因组dna片段修整成平末端。在某些示例性实施方案中,所述末端修复过程包括3’末端悬突的切除过程和5’末端悬突的补平过程。在某些优选的实施方案中,所述末端修复过程可利用含有3’至5’外切活性的核酸聚合酶进行。
204.第二步,经过体外标记方法在碱基编辑中间体中编辑碱基(例如次黄嘌呤)所在的位置下游掺入经第一标记分子(例如生物素)标记的核苷酸(例如尿嘧啶脱氧核糖核苷酸)。在某些示例性方案中,所述标记实验包括:使用核酸内切酶endo v对碱基编辑中间体中的次黄嘌呤进行特异性识别,并切割次黄嘌呤脱氧核糖核苷酸3’端第二个磷酸二酯键,形成单链缺口;利用含有链置换活性的dna聚合酶从产生的单链缺口开始沿着5’至3’方向进行dna链置换反应;用dna连接酶连接dna链置换反应产物中的单链切口。其中,所述dna链置换反应体系中,使用至少一种经第一标记分子(例如生物素)标记的核苷酸底物(例如生物素-尿嘧啶核糖核苷酸)来代替常规的核苷酸酸底物(例如胸腺嘧啶脱氧核糖核苷酸)。经第一标记分子标记的核苷酸(例如生物素-尿嘧啶脱氧核糖核苷酸)的掺入可以使得后续能够利用所述第一结合分子(例如链霉亲和素)富集含有第一标记分子的dna片段。碱基编辑中间体中含有的编辑碱基(例如次黄嘌呤)在后续dna复制和测序过程中会与胞嘧啶互补配对,从而,在标记产物的测序结果中,次黄嘌呤的位置会产生a-to-g的突变信号。由此,通过检测突变信号的存在,可以实现对编辑碱基(例如,次黄嘌呤)所在位置的精准定位。
205.在某些优选的实施方案中,为避免内源性或核酸操作过程中引入的dna损伤(例如,ssb)可能带来的假阳性信号,在进行第二步之前,所述方法还包括,对编辑产物进行核酸修复处理。在某些示例性实施方案中,所述处理包括:用dna聚合酶从ssb缺口处开始沿着5’至3’方向进行dna链置换反应;用dna连接酶连接链置换反应产物中的缺口。在某些优选的实施方案中,所述dna聚合酶具有链置换活性。
206.第三步,利用偶联有第一结合分子(例如链霉亲和素)的固相支持物(例如磁珠)富集含有第一标记分子(例如生物素)的dna片段;其任选地经过扩增和/或文库构建后,可用于高通量测序。根据测序结果,可分析腺嘌呤碱基编辑器编辑靶核酸后产生的碱基编辑中间体(例如含有次黄嘌呤的dna)中编辑位点的位置信息。
207.在某些优选的实施方案中,在对富集的dna片段进行扩增和/或文库构建前,还可以对固相支持物(例如磁珠)上富集的dna片段进行处理(例如碱处理),以去除含有第一标记分子(例如生物素)的核酸单链的互补链。
208.在某些示例性实施方案中,在用碱(例如naoh)处理以去除含有第一标记分子(例如生物素)的核酸单链的互补链之前,通过接头连接反应在富集的dna片段末端连上寡核苷酸接头,以便于dna片段的扩增或测序。在某些优选的实施方案中,在dna片段的3’端添加da尾,所述da尾可用于与含有dt尾的寡核苷酸接头连接。
209.图16显示了本发明实施例2的方法对不同模式序列的富集结果。
210.图17显示了各样品组abe在hek293_site_4sgrna(简称为hek4)的靶向位点处的高通量测序结果。阴影指示on-target所在的序列位置,其中“g”即为a-to-g的突变信号。
211.图18显示了各样品组abe在hek4的一个脱靶位点(off-target 4)处的高通量测序结果。阴影指示sgrna可能结合的序列位置,其中“g”即为a-to-g的突变信号。
212.图19显示了abe在hek4的脱靶位点(off-target 4)的定点深度测序验证结果。前两行序列分别是on-target的序列和脱靶位点的序列;最后六行代表a、g、c、t碱基及插入(insertion)、缺失(deletion)所占的比例。
213.图20显示了hek4 sgrna在abe,abe8e和acbe系统中的靶向编辑位点处高通量测序结果。橙色g代表a-to-g突变信号;红色t代表c-to-t突变信号。
214.图21显示了hek4 sgrna在abe,abe8e和acbe系统中的脱靶位点(off-target4)处的高通量测序结果。橙色g代表a-to-g突变信号;红色t代表c-to-t突变信号。
215.图22显示了abe,abe8e和acbe系统在abe8e-only脱靶位点处的高通量测序结果。蓝色c代表t-to-c突变信号,亦即代表其互补链上的a-to-g突变信号。
216.图23显示了将本发明中丙二腈标记步骤替换为其他5fc标记法(吡啶硼烷标记反应或2-甲基吡啶硼烷标记反应)后,本发明对spike-in序列上的表征结果。其中,(图23a)为替换为吡啶硼烷等(吡啶硼烷或2-甲基吡啶硼烷)化学标记法后本发明对不同模式序列(ap:da、du:da或du:dg)的qpcr富集结果;(图23b)为替换为吡啶硼烷等(吡啶硼烷或2-甲基吡啶硼烷)化学标记法后本发明对含du:dg碱基对模式序列的sanger测序结果。红色箭头指示化学标记引发的c-to-t突变信号。
217.图24显示了将本发明中的biotin-du替换为biotin-dg后本发明对不同模式序列(nick、ap:da、du:da或du:dg)的qpcr富集结果。
218.序列信息
219.本发明涉及的序列的信息提供于下面的表1中。
220.表1
221.222.[0223][0224]
注:符号“^”表示nick位点;n=a,t,g,or c;符号“p”表示磷酸化修饰;“amn”表示c7 aminolinker封闭。
具体实施方式
[0225]
现参照下列意在举例说明本发明(而非限定本发明)的实施例来描述本发明。
[0226]
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。本领域技术人员知晓,实施例以举例方式描述本发明,且不意欲限制本技术所要求保护的范围。
[0227]
实施例1:cbe编辑位点检测
[0228]
实验方法:
[0229]
1.dna片段化
[0230]
提取经cbe系统转染的hek293t(购自atcc,货号:crl-11268)或mcf7(购自atcc,货号:htb-22)活细胞基因组dna。cbe系统转染细胞的方法参见(xiao wang,et al.nature biotechnology 36,946-949,doi:10.1038/nbt.4198(2018)),细胞基因组dna提取方法参见试剂盒说明书(购自康为世纪,货号:cw2298m)。
[0231]
将提取的基因组dna通过covaris me220超声破碎仪打断至~300bp左右长度的片段,随后通过dna clean&concentrator-5kit(购自vistech,货号:dc2005)进行回收。
[0232]
2.dna片段末端修复
[0233]
按上述步骤1进行片段化后的dna会有一些切口(nick)以及末端突出(overhangs),这些如果不被修复掉会在后续的标记反应中被标记上biotin从而产生假阳
性。故而本步骤使用neb末端修复模块(货号:e6050)及e.coli dna ligase(购自neb,货号:m0205)来修复打断过程可能造成的基因组dna损伤。
[0234]
按照表2配制反应体系:
[0235]
表2:末端修复反应体系
[0236][0237]
将上述反应体系在冰上混匀后,于20℃反应30min,之后用2.0
×
ampure xp beads(购自beckman coulter,货号:nc9933872)回收,ddh2o洗脱。
[0238]
3.etonh2保护
[0239]
将步骤2制备的经末端修复的dna片段在80μl含有10mm etonh2的100mm mes缓冲液(ph 5.0)中于37℃孵育6h,使得细胞内天然存在的d5fc修饰被保护住而无法与后续使用的丙二腈反应产生假阳性。随后,使用dna clean&concentrator-5kit回收反应后的dna。
[0240]
4.加da尾
[0241]
将步骤3所得dna片段3’末端各添加上一个da,以方便后续利用a/t互补规则连接上测序接头(adaptor)。
[0242]
按照表3配制反应体系:
[0243]
表3:加da尾反应体系
[0244][0245]
将上述反应体系在冰上混匀后于37℃反应30min,之后用2.0
×
ampure xp beads回收,ddh2o洗脱。
[0246]
5.dna损伤修复
[0247]
此步骤目的是为了把细胞内天然存在的ap位点、ssb、nick等可能产生假阳性信号的dna修饰或损伤在du标记之前进行修复去除。
[0248]
按照表4配制反应体系:
[0249]
表4:损伤修复反应体系
[0250]
组分总体系(50μl)经步骤4制备的dna38μl(~2.7ug)nebuffer 3.0(购自neb,货号:b7003s)5μl
50mm的nad
+
1μl2.5mm dntps1μlendo iv(购自neb,货号:m0304)2μlbst full-length polymerase(购自neb,货号:m0328)1μltaq dna ligase(购自neb,货号:m0208)2μl
[0251]
将上述反应体系混匀后先在37℃反应60min,之后45℃反应60min。用2.0
×
ampure xp beads回收,ddh2o洗脱。
[0252]
6.体外ber标记试验
[0253]
将上述步骤5得到的dna留取0.5μl再加0.5μl ddh2o作为input,剩余样品按如下步骤进行标记反应。
[0254]
按照表5配制反应体系:
[0255]
表5:体外标记反应体系
[0256]
组分总体系(50μl)经步骤5制备的dna37μl(~2.5ug)nebuffer 3.05μl50mm的nad
+
1μl5μm datp/dgtp/biotin-dutp/20μm d5fctp2μludg(购自neb,货号:m0280)1μlendo iv1.5μlbst full-length polymerase0.8μltaq dna ligase1.7μl
[0257]
将上述反应体系混匀后置于37℃反应40min,用2.0
×
ampure xp beads回收,ddh2o洗脱。
[0258]
7.丙二腈反应
[0259]
将上述步骤6回收所得dna置于含有75mm malononitrile(丙二腈)的50mm tris-hcl(ph 7.0)中,并置于37℃,转速800rpm的混匀仪(mixer)中反应20h。随后再次通过2
×
ampure xp beads进行回收,ddh2o洗脱。
[0260]
8.片段富集
[0261]
每一个pd(pull down)样品对应10μl streptavidin c1 beads(购自invitrogen,货号:65002)。取足量的beads用1
×
b&w buffer(5mm tris-hcl(ph 7.5),1m nacl,0.5mm edta,0.05%tween-20)清洗3次后,用40μl 2
×
b&w buffer重悬,再加入等体积的经上述步骤7处理的样品dna,混匀后置于室温旋转孵育1h。而后用1
×
b&w buffer清洗磁珠3次,再用10mm tris-hcl(ph 8.0)清洗1次,每次置于室温旋转5min。最后,在磁力架上将tris-hcl液体吸出,将剩下的结合有dna片段的磁珠(体积大约为1μl)用于接头连接反应。
[0262]
9.连接接头
[0263]
1)用10mm tris-hcl在冰上将adaptor储液(30μm)稀释至1.5μm。所用y型adaptor由两条单链序列进行退火反应而得,其中,正向单链5’端带有磷酸化修饰,且3’端被一个c7 aminolinker封闭,其序列如seq id no:7所示,反向单链序列如seq id no:8所示。
[0264]
2)使用quick ligation module(购自neb,货号:e6056)对步骤6留
存的input样品(水溶液)及上述步骤8所得的pd样品(连接于磁珠上)做接头连接反应。
[0265]
按照表6配制反应体系:
[0266]
表6:接头连接反应体系
[0267]
组分总体系(25μl)ddh2o14μlneb quick ligation buffer5μl1.5μm y型adaptor2.5μlquick t4 dna ligase2.5μlpd或input样品dna1μl
[0268]
对于pd样品的接头连接反应:将上述反应体系混匀后置于约20℃旋转反应(避免磁珠沉降)1h,随后补加50μl 1
×
b&w buffer,继续室温旋转孵育1h(使在连接过程脱离下来的少量dna片段重新与磁珠结合),而后进行下一步反应;
[0269]
对于input样品的接头连接反应:将上述反应体系混匀后置于pcr仪20℃反应40min,使用1
×
ampure xp beads进行回收留存,以去除未连接成功的adaptor。
[0270]
10.naoh处理
[0271]
对上述步骤9获得的磁珠上的pd样品,用1
×
b&w buffer清洗3次,再用1
×
ssc buffer清洗1次,每次先轻轻颠倒将磁珠荡起而后置于室温旋转5min。而后去除上清,将剩余的磁珠重悬于20μl 0.15m naoh溶液并置于室温旋转孵育10min,再用1
×
ssc buffer、10mm tris-hcl(ph 8.0)接连清洗1次。最后用ddh2o于95℃处理磁珠3min,将磁珠上的dna文库洗脱下来用于下一步pcr扩增。
[0272]
11.文库扩增
[0273]
1)因高保真dna聚合酶的扩增过程易被biotin-du与标记了丙二腈的d5fc所截断,故而先使用保真性稍低的mightyamp dna polymerase(购自takara,货号:r076a)对文库进行扩增。
[0274]
按照表7配制反应体系:
[0275]
表7:mightyamp扩增体系
[0276][0277][0278]
将上述反应体系混匀后进行pcr反应。程序为:98℃ 30s;98℃ 10s,65℃ 90s(2个循环);72℃ 5min。使用dna clean&concentrator-5kit(vistech)回收反应后的dna。
[0279]
2)使用高保真dna聚合酶进行后续扩增以保证较低的整体测序噪音背景。
[0280]
按照表8配制反应体系:
[0281]
表8:高保真扩增体系
[0282][0283]
将上述反应体系混匀后进行pcr反应。程序为:98℃ 30s;98℃ 10s,65℃ 90s(pd样品8-9个循环;input样品6-7个循环);72℃ 5min。用0.9
×
ampure xp beads回收pcr产物,ddh2o洗脱。
[0284]
12.文库质检
[0285]
用qubit2.0精密分光光度计测定文库浓度;
[0286]
用fragment analyzer 12全自动毛细管电泳仪检查文库片段分布;
[0287]
用qpcr对模式序列进行相对定量并计算富集倍数,qpcr所用引物如seq id nos:11-22所示,数据处理采用2
‑△△
ct
法,富集倍数即为含有特定类型修饰的spike-in dna分子在pd样品中的相对量(以control模式序列为参考)相比于对应input样品的变化倍数,基于此倍数可评估本批实验的富集情况;
[0288]
对模式序列进行全长pcr扩增,用所得pcr产物进行sanger测序,通过测序结果可评估本批实验的标记情况;
[0289]
最后将所得文库递送illumina hiseq x-ten平台进行双端测序(读长150bp)。
[0290]
测序数据处理与分析:
[0291]
1.本发明数据的回贴与过滤
[0292]
数据下机后,首先使用cutadapt(version 1.18)软件对测序结果的fastq文件中的测序读段(reads)进行测序接头的去除,具体命令参数为:cutadapt
‑‑
times 1-e 0.1-o 3
‑‑
quality-cutoff 25-m 50。去除接头以后,考虑到本发明测序结果中会包含c到t的突变,因此首先使用bismark(version 0.22.3)软件将去除测序接头的测序读段回贴到参考基因组(版本号为hg38)。没有比对成功或比对质量maqp低于20的测序读段会被重新提取出来,然后再使用bwa mem(version 0.7.17)进行重新比对。最终经过两次比对合并后的测序数据会被再次筛选,只有比对质量mapq大于20,即低于1%比对错率的比对结果才会被保留进行下游分析。接下来,对筛选的高质量比对结果进行去重复处理,使用picard markduplicates命令(version 1.9)进行操作,这一步主要目的是去除文库构建过程中由于扩增产生的分子冗余。经过上述步骤,即可获得可供下游分析的基因组回贴结果(bam格式文件)。
[0293]
2.本发明信号的初步鉴定
[0294]
使用samtools mpileup-q 20-q 20命令(version 1.9)将bam文件转换成mpileup文件。随后,使用编写的软件工具(参见例如,https://github.com/menghaowei/detect-seq)中的parse-mpileup命令及bmat2pmat命令生成pmat文件。接着再使用pmat-merge命令对全基因组所有串联的c到t突变信号进行扫描整理并记录成mpmat格式文件。最后使用mpmat-select命令进行筛选,获得初步的本发明测序信号。
[0295]
3.本发明富集信号的鉴定
[0296]
在获得初步的本发明测序信号后,需对这些候选区域进行富集检测。首先使用所述软件工具中的find-significant-mpmat命令,对候选区域进行统计检验,统计检验的结果会经过bh方法进行矫正得到假发现率(fdr)。最终认为fdr小于0.01,处理组相比对对照组归一化后的富集倍数大于2,对照组样本中带有突变信号的读段小于3,处理组样本中带有突变信号测序读段不小于5的区域为本发明最终鉴定区域。
[0297]
4.内源脱氧尿嘧啶位点的去除
[0298]
在富集检测中,将实验组和对照组分别设置为只转染了空载质粒并进行本方法描述的富集建库流程的样本和未经本方法描述的富集建库流程处理的样本,即可获得内源脱氧尿嘧啶的位置信息。为了保证此鉴定方法具有更低的假阴性率,在这一步使用较为宽松的阈值:fdr小于0.05,实验组相比对照组归一化后的富集倍数大于1.5。
[0299]
5.脱靶位点基因序列与sgrna序列的比对
[0300]
在上述步骤鉴定到的去除内源du的富集信号区域中,可以通过序列比对的方法推测sgrna/crrna的结合位置。此推测出的sgrna/crrna结合位置被称为prbs(putative sgrna/crrna binding site)。在进行sgrna/crrna与富集信号区域内进行序列比对时,使用了改进后的半-全局比对(semi-global alignment)的方法。对于sgrna,首先会在区域内搜索pam序列(nag/ngg),随后对于找到的pam位置,会提取pam 5’方向30nt的序列与sgrna进行半-全局双序列比对,比对中报告的最优结果即为prbs;对于crrna,首先会在区域内搜索pam序列(tttv,v=a/c/g),随后在搜索到的pam位点,提取pam 3’方向30nt的序列与crrna进行半-全局双序列比对,比对中报告的最优结果即为crrna的prbs。上述过程中,若在区域内未发现pam,则直接将sgrna/crrna与该区域序列进行半-全局比对,比对最优结果为该sgrna/crrna的prbs。该步骤使用的比对参数为匹配+5;不匹配-4;打开间隔-24;间隔延伸-8。此步骤的比对程序包含在detect-seq软件工具箱中的mpmat-to-art命令。
[0301]
实验结果:
[0302]
1.含du的模式序列的特异性标记与富集
[0303]
为了证明本发明的方法的特异性和效率,将图2a所示的含有不同修饰碱基的模式序列和对照序列(seq id nos:1-6)掺入至打断后的基因组dna中,再按上述实验方法进行建库。最后通过荧光定量pcr技术计算和比较了pull-down前后样品中不同模式序列的比例变化(均与不含任何修饰的对照序列(seq id no:1所示的control模式序列)进行相对定量),并计算pull-down前后样品中不同模式序列的富集倍数。富集倍数如图2b所示,由图可知,对于含有单个du:da和du:dg碱基对的模式序列,本发明提供的方法可以将之分别富集约60倍和约30倍;而对于含有ap位点、d5fc的模式序列则几乎完全没有富集。说明本发明提供的方法可以特异性地富集含du的dna片段。
[0304]
另一方面,按照原理设计,本发明一定概率会在du所在位置的3’端连续性地掺入多个d5fctp,从而使得其后产生连续性的c-to-t突变,以此实现信号放大而利于检测的目的。从sanger测序以及高通量测序的结果(图3),我们也确实在含du的模式序列上观察到了连续性的c-to-t突变信号,说明本发明流程中通过化学反应引入c-to-t突变信号的策略确实可以实现du位置的标记。
[0305]
综上,通过对这种极具特点的c-to-t突变信号的捕捉,可以实现非常灵敏和准确的du检测。
[0306]
2.cbe编辑位点处产生特异性的检测信号
[0307]
在人类hek293t和mcf7细胞系中,挑选几个具有代表性的sgrna用于试验本发明提供的方法对高效cbe工具be4max脱靶效应的检测。cbe4max编辑系统转染细胞的方法参见(xiao wang,et al.nature biotechnology 36,946-949,doi:10.1038/nbt.4198(2018))。所述具有代表性的sgrna分别为已知在体内特异性很低的“vegfa_site_2”(seq id no:23)与“hek293 site_4”(seq id no:24)、特异性中等的“emx1”(seq id no:25)、未被报道过有脱靶位点的“rnf2”(seq id no:26)以及此前研究较少的“runx1”(seq id no:27)。
[0308]
检测结果如图4所示,由图4a可知,本发明的方法在对应on-target编辑位点处造成了一个非常明显的reads富集峰(peak),进一步放大后可以观察到明显的、特征性的连续性c-to-t突变信号;并且,这些富集突变信号在作为阴性对照的nt样品(即转染了be4max与non-target sgrna的样品)中并没有被观察到,说明本发明具有非常好的检测特异性。通过与此前研究中这些sgrna的on-target编辑结果相对比,我们发现这些c-to-t突变信号最强的c通常就是真实编辑效率最高的胞嘧啶位置。而且可能是因为本发明中的聚合酶切口平移反应可以一次性掺入多个d5fctp,即使是只有一两个c被编辑也会产生明显的连续性c-to-t突变信号。由图4b可知,一般主要会在被编辑的c后4-9bp区域内产生2-6个连续的c-to-t突变。
[0309]
此外,以图4c为例,可以明显看出本发明产生的连续性c-to-t突变特征性信号可以非常容易地与snv进行区分。并且从全基因组水平来看,本发明的方法在相同的数据量情况下所产生的信号要远远强于常规wgs测序,远比之更容易与测序本底误差进行区分,对测序覆盖度的要求更低(图4d)。
[0310]
综上,以上观察说明本发明的方法产生的信号特征可以大大增强编辑位点处的检测信号,从而大大提高本发明的检测灵敏度,降低检测成本。
[0311]
3.cbe造成的cas依赖型脱靶与非cas依赖型脱靶的评估
[0312]
通过对cbe系统做不同组分的删除对比实验,可验证本发明在全基因组水平检测到的脱靶位点性质及其可能的产生机制。具体地,我们在转染细胞时分别将be4max系统中的apobec1、ugi和sgrna部分分别进行了去除,去除后各质粒构成如图5所示,同时使用了只转染mcherry质粒的vector样品作为阴性对照样品,再分别对这些样品转染后的基因组dna使用本发明的方法进行检测。
[0313]
非cas依赖型脱靶的检测结果如图6所示,其呈现出三个明显的特征:1)信号所在的基因位置与sgrna序列几乎没有相似性(图6a);2)通常信号强度非常低,大多刚刚超过本底水平(图6a);3)更倾向于出现在转录活跃区域(图6e)。这些特征与此前报道的非cas依赖型脱靶表现一致。更重要的是,当对这类脱靶位点进一步分析时,可以看到:当cbe系统所有元件都齐全时,找到的此类脱靶位点数目较多,且展现出一个非常明显的“tc”基序(tc motif);当去除了sgrna组分后,此类位点数目依然很多,且该基序依然存在;但删除了apobec1组分后,此类位点的数目即降为本底,且基序也随之消失了(图6b-d)。已知apobec1对于“tc”基序具有天然的底物结合偏好性。这些实验数据和特征说明此类脱靶位点并不依赖于cas系统,而是仅依赖于apobec1产生,应为由apobec1过表达而随机产生的脱靶编辑。
[0314]
cas依赖型脱靶的检测结果如图7所示,其表现出以下特征:1)大部分信号强度要比非cas依赖型脱靶强得多。在某些位点甚至可以观察到堪比on-target位点处的信号强度
(图7a),指示着此类脱靶位点的编辑效率会高得多;2)在生物学重复组中重复稳定地产生信号(图7b);3)在信号所在的基因组区域通常可以找到与sgrna具有一定相似性的基因序列。通过组分删除对比实验可以看到:相较于所有元件齐全的all样品,(-)sgrna样品和(-)apobec样品中此类位点的信号强度全都降至本底背景水平以下,(-)ugi样品中的信号强度减弱程度不一;而细胞内源存在的du修饰位点信号强度则几乎完全不受到组分删除的影响(图7c)。这些实验数据表明此类脱靶位点应是同时依赖于sgrna与apobec而产生,应确为经典的cas依赖型脱靶。此外,对于特异性不同的sgrna,本发明鉴定到的cas依赖型脱靶位点的个数也会随之改变:比如在相同的生信分析鉴定规则(cufoff)下,对于已知特异性非常差的“vegfa_site_2”,本发明一共鉴定到了511个此类脱靶位点(图7b);而对于已知特异性极好的“rnf2”,本发明则没有检测到此类脱靶位点。
[0315]
4.脱靶位点的验证结果
[0316]
为了验证本发明的方法检测结果的真实性,采用定点深度测序(targeted deep sequencing)技术衡量本发明鉴定到的脱靶位点处的实际编辑效率。所谓定点深度测序技术即是对目标待测位点进行定点pcr扩增,然后再对其pcr产物进行高通量测序,从而可使得被测基因组位点处覆盖至少上万reads的测序深度,故而可以获得此位点非常精准的编辑效率。
[0317]
采用定点深度测序对本发明的方法检测到的位点进行验证的结果如图8所示,由图可知,随机挑选的本发明信号强度从低到高的位点(总共151个)中,50/50个“emx1”位点、51/51个“vegfa_site_2”位点、43/43个“hek293site_4”和7/7个“runx1”位点均被深度测序方法成功验证,具有高达近100%的真阳性率。并且,当实际编辑效率还处于较低水平的时候,对应的本发明信号强度却已经很高,这进一步说明了本发明确实具有非常高的检测灵敏度。
[0318]
此外,通过定点深度测序法验证了本发明的方法鉴定到的cas依赖型脱靶(共选取了20多个位点)确实是依赖于sgrna产生,图9示出了在有无sgrna的样品组在其中两个位点的深度测序信号,图9的结果表明所述两处脱靶位点确实是依赖于sgrna产生,综上,以上数据证明了本发明的方法的高可信度。
[0319]
图10显示了利用本发明的方法在全基因组水平检测到的“emx1”、“vegfa_site_2”与“hek293 site_4”sgrna靶向编辑位点和cas依赖型脱靶编辑位点在各染色体上的分布。
[0320]
5.本发明的方法(detect-seq)与其他相关方法检测结果的比较
[0321]
guide-seq是一项为基因编辑领域广泛熟知的脱靶检测技术,主要用于检测crispr/cas9核酸酶系统造成的cas依赖型脱靶。鉴于cbe工具也是基于失活或部分失活的cas9蛋白而构建,于是部分学者便直接通过guide-seq鉴定到的位点来评估cbe系统的脱靶效应。但实际上,即使是使用相同的sgrna,cbe系统造成的全基因组脱靶和cas9核酸酶造成的脱靶还是非常不一样的(kim,d.et al.nature biotechnology 35,475-480,doi:10.1038/nbt.3852(2017).)。
[0322]
本发明的方法与guide-seq检测结果的比较如图11a所示,对于“vegfa_site_2”与“emx1”,本发明的方法检测到了guide-seq结果中的大部分cas依赖型脱靶位点;对于“hek293 site_4”,本发明方法则检测到guide-seq的约一半位点;本发明方法新发现了非常多guide-seq未曾报道过的脱靶位点。随机挑点进行定点深度测序验证的结果表明:相比
guide-seq,本发明方法检测到的41个新脱靶位点确实均为真实的脱靶位点,而15/17个本发明方法未报告却被guide-seq报道的位点处在活细胞内确实未发生cbe编辑事件;37个被两者一起鉴定到的脱靶位点均被验证成功。
[0323]
本发明的方法与kim等人研发的针对cbe系统的digenome-seq检测结果的比较如图11b所示,digenome-seq本质上是一种基于wgs建立的体外脱靶检测技术。类似于同常规wgs比较的结果,相同测序量情况下本发明在脱靶位点处展现出的信号值要远远高于digenome-seq。本发明的方法检测到了digenome-seq报道的大部分cas依赖型脱靶位点,但新发现了个数远多于后者的脱靶位点(图11b)。随机挑点进行定点深度测序验证的结果表明:10/15个本发明未报告却被digenome-seq报道的位点处在活细胞内确实未发生cbe编辑事件;18个被两者一起鉴定到的脱靶位点均被验证成功。
[0324]
以上结果另一方面也表明本发明的报道的真阳性率接近100%,而真阴性率大约为80%。值得一提的是,如若进一步仔细查验本发明的方法的检测结果,其实也可以在未被成功报道的7个真实脱靶位点处观察到程度高低不同的检测信号,但可能因未达到生信分析的阈值(cutoff)而未被报道。
[0325]
6.优化版cbe工具脱靶效应的评估
[0326]
近期本领域内报道了很多在降低dna或rna脱靶效应方面表现优秀的cbe改进工具,其中以ye1-be4max被多项独立研究报道为综合最优的cbe版本(doman,et al.nature biotechnology 38,620-628,doi:10.1038/s41587-020-0414-6(2020);zuo,e.et al.nat methods 17,600-604,doi:10.1038/s41592-020-0832-x(2020))。
[0327]
通过本发明的方法可检测到ye1-be4max确实降低了wt-be4max造成的大部分脱靶信号水平。然而,以“emx1”sgrna为例,从wt-be4max样品鉴定到的48个cas依赖型脱靶位点中,依然还有4、3、十几个位点在ye1-be4max中保留有高、中、低强度的检测信号(图12a)。
[0328]
定点深度测序的验证结果表明:在on-target位点编辑效率差不多的情况下,ye1-be4max确实在本发明报告阴性的位点(例如“emx1 prbs_1”位点)基本不产生编辑结果;而在本发明鉴定到的3个强信号位点处(“emx1prbs_4”emx1 prbs_3”emx1 prbs_2”位点),ye1-be4max依然表现出了非常高的脱靶编辑比例(最高可高达近乎on-target编辑效率的一半),在其中一个位点(emx1 prbs_2”位点)更是相比于wt-be4max完全没有降低。可见,用本发明评估新优化工具的整体脱靶效应具有较高的可信度。并且同理,其他优化版本的cbe工具(比如使用apobec3a构建的cbe系统)亦可通过本发明进行综合脱靶评估。
[0329]
此外,这些数据另一方面也说明:此前仅通过随机挑选guide-seq鉴定到的部分位点来进行cbe工具的脱靶效应评估还是不够全面,得到的结论很可能因挑选的位点不同而不同。而本发明可以提供一个基于全基因组水平综合考量的评估平台,为cbe工具的优化和比较提供考量依据。
[0330]
7.基于其他crispr系统构建的cbe工具脱靶的检测
[0331]
鉴于相同的apobec脱氨编辑原理,基于其他crispr系统构建的cbe工具,比如cpf1(cas12a)-be,亦可使用本发明的方法进行脱靶评估。图13显示了利用本发明的方法对“runx1”(seq id no:37)与“dyrk1a”(seq id no:38)crrna由lbcpf1-be在全基因组水平造成的949和240个cas依赖型脱靶位点。同样地,定点深度测序验证了其中18/18是真实的脱靶编辑位点。
[0332]
8.crispr-free的ddcbe工具的脱靶检测
[0333]
用分别靶向线粒体不同dna位点的ddcbe系统转染hek293t细胞,转染方法参见(mok,b.y.et al.nature 583,631-+,doi:10.1038/s41586-020-2477-4(2020))。三天之后提取基因组检测线粒体靶向位点处的编辑效率,sanger测序结果显示其编辑效率在35%-55%之间。鉴于ddcbe系统中的脱氨酶ddda会将双链dna上的dc转变成du,因此也可用本发明的方法检测中间产物du,进而评估ddcbe造成的脱靶。
[0334]
尽管ddcbe是线粒体dna胞嘧啶编辑工具,但detect-seq的结果显示,每种ddcbe在细胞核中都有数百个脱靶编辑。根据脱靶信号的特征以及产生原因,可将脱靶信号分为两大类,分别为tale依赖型脱靶和非tale依赖型脱靶。本发明中随机选取了36个脱靶位点进行验证,定点深度测序结果证实这36个位点确实都存在一定的脱靶编辑比例,有的位点脱靶效率甚至高达8%,说明detect-seq的确可以用于检测ddcbe造成的脱靶。图14示例性示出了本发明的方法检测到的tale依赖型脱靶和非tale依赖型脱靶的测序信号图以及采用定点深度测序对其进行验证的测序结果。
[0335]
实施例2:abe编辑位点检测
[0336]
实验方法:
[0337]
1.dna片段化
[0338]
提取经abe系统转染的hek293t(购自atcc,货号:crl-11268)活细胞基因组dna。abe系统转染细胞的方法参见(xiao wang,et al.nature biotechnology 36,946-949,doi:10.1038/nbt.4198(2018)),细胞基因组dna提取方法参见试剂盒说明书(购自康为世纪,货号:cw2298m)。
[0339]
将提取的基因组dna通过covaris me220超声破碎仪打断至~300bp左右长度的片段,随后通过dna clean&concentrator-5kit进行回收。
[0340]
2.dna片段末端修复
[0341]
本步骤使用neb末端修复模块和e.coli dna ligase来补平片段化dna的一些切口(nick)和末端突出(overhangs),以及修复打断过程可能造成的基因组dna损伤。
[0342]
按照表9配制反应体系:
[0343]
表9:末端修复反应体系
[0344][0345]
将上述反应体系在冰上混匀后,于20℃反应30min,之后用2.0
×
ampure xp beads回收,40μl ddh2o洗脱。
[0346]
3.加da尾
[0347]
将步骤2所得dna片段3’末端各添加上一个da,以方便后续利用a/t互补规则连接上测序接头(adaptor)。实验步骤同实施例1。
[0348]
4.dna损伤修复
[0349]
按照表10配制反应体系:
[0350]
表10:损伤修复反应体系
[0351]
组分总体系(50μl)经步骤3制备的dna40μl(~3.3μg)nebuffer 3.05μl50mm的nad
+
1μl2.5mm dntps1μlbst full-length polymerase1μltaq dna ligase2μl
[0352]
将上述反应体系混匀后先在37℃反应60min,之后45℃反应60min。用2.0
×
ampure xp beads回收,用17μl ddh2o洗脱,取1μl样品作为input后续建库备用。
[0353]
5.di识别
[0354]
本步骤的目的是为了使在di 3’端第二个磷酸二酯键断裂,从而产生一个切口,以便后续的标记。
[0355]
按照表11配制反应体系:
[0356]
表11:切口形成反应体系
[0357]
组分总体系(20μl)经步骤4制备的dna16μl(~3μg)nebuffer 42μlendonuclease v(购自neb,货号:m0305)2μl
[0358]
将上述反应体系混匀后,37℃反应80min,之后用两倍体积xp beads进行纯化,最后用43μl水洗脱。
[0359]
6.biotin-标记
[0360]
本步骤的目的是为了在需要检测的位置加入biotin标记的dutp。
[0361]
按照表12配制反应体系:
[0362]
表12:biotin-标记反应体系
[0363]
组分总体系(50μl)经步骤5制备的dna42μl(~2.7μg)nebuffer 35μl100mm datp0.5μl100mm dctp0.5μl100mm dgtp0.5μl5μm biotin-16-aa-2
’‑
dutp0.5μlfull length bst dna polymerase1μl
[0364]
将上述反应体系混匀后,37℃反应40min,反应结束后再向管中加入1μl 50mm nad
+
和2μl taq dna ligase,且继续在pcr仪器中37℃孵育40min,反应结束后用2
×
xp beads进行纯化,最后用41μl水洗脱。
[0365]
7.片段富集
[0366]
每一个pd(pull down)样品对应10μl streptavidin c1 beads。取足量的beads用1
×
b&w buffer(5mm tris-hcl(ph 7.5),1m nacl,0.5mm edta,0.05%tween-20)清洗3次后,用40μl 2
×
b&w buffer重悬,再加入等体积的经上述步骤6处理的样品dna,混匀后置于室温旋转孵育1h。而后用1
×
b&w buffer清洗磁珠3次,再用10mm tris-hcl(ph 8.0)清洗1次,每次置于室温旋转5min。最后,在磁力架上将tris-hcl液体吸出,将剩下的结合有dna片段的磁珠用于接头连接反应。
[0367]
8.连接接头
[0368]
1)用10mm tris-hcl在冰上将adaptor储液(30μm)稀释至1.5μm。所用y型adaptor由两条单链序列进行退火反应而得,其中,正向单链5’端带有磷酸化修饰,其序列如seq id no:7所示,反向单链序列如seq id no:8所示。
[0369]
2)使用quick ligation module对步骤4留存的input样品(水溶液)及上述步骤7所得的pd样品(连接于磁珠上)做接头连接反应。
[0370]
按照表13配制反应体系:
[0371]
表13:接头连接反应体系
[0372]
组分总体系(25μl)ddh2o14μlneb quick ligation buffer5μl1.5μm y型adaptor2.5μlquick t4 dna ligase2.5μlpd或input样品dna1μl
[0373]
对于pd样品的接头连接反应:将上述反应体系混匀后置于约20℃旋转反应(避免磁珠沉降)1h,随后补加50μl 1
×
b&w buffer,继续室温旋转孵育1h(使在连接过程脱离下来的少量dna片段重新与磁珠结合),而后进行下一步反应;
[0374]
对于input样品的接头连接反应:将上述反应体系混匀后置于pcr仪20℃反应1h,使用1
×
ampure xp beads进行回收留存,以去除未连接成功的adaptor。
[0375]
9.清洗纯化过程
[0376]
对上述步骤8处理后连接在beads上的样品(pd样品)用1ml 1
×
bw清洗三次,随后用200μl eb(10mm tris-hcl)清洗一次,最后用25μlddh2o在95℃1200rpm的条件的shaker中洗脱出pd样品中的dna文库。
[0377]
10.文库扩增
[0378]
实验步骤同实施例1。
[0379]
11.文库质检
[0380]
用qubit2.0精密分光光度计测定文库浓度;
[0381]
用fragment analyzer 12全自动毛细管电泳仪检查文库片段分布;
[0382]
用qpcr对模式序列进行相对定量并计算富集倍数,qpcr所用引物如seq id nos:11-12,31-36所示,数据处理采用2
‑△△
ct
法,富集倍数即为含有特定类型修饰的spike-in dna分子在pd样品中的相对量(以control模式序列为参考)相比于对应input样品的变化倍数,基于此倍数可评估本批实验的富集情况;
[0383]
对模式序列进行全长pcr扩增,用所得pcr产物进行sanger测序,通过测序结果可评估本批实验的标记情况;
[0384]
最后将所得文库递送illumina hiseq x-ten平台进行双端测序(读长150bp)。
[0385]
测序数据处理与分析:
[0386]
1.本发明数据的回贴与过滤
[0387]
数据下机后,首先使用cutadapt(version 1.18)软件对测序结果的fastq文件中的测序读段(reads)进行测序接头的去除,具体命令参数为:cutadapt
‑‑
times 1-e 0.1-o 3
‑‑
quality-cutoff 25-m 50。去除接头以后的测序读段使用bwa mem(version 0.7.17)进行回贴到参考基因组(版本号为hg38),比对质量mapq大于20,即低于1%比对错率的比对结果会被保留进行下游分析。随后使用picard markduplicates命令(version 1.9),对筛选的高质量比对结果进行去重复处理,这一步主要目的是去除文库构建过程中由于扩增产生的分子冗余。经过上述步骤,即可获得可供下游分析的基因组回贴结果(bam格式文件)。
[0388]
2.本发明信号的初步鉴定
[0389]
在获得回贴过滤好的bam文件后,首先使用samtools mpileup-q 20-q20命令(version 1.9)将bam文件转换成mpileup文件。随后,使用上文所述软件工具中的parse-mpileup命令及bmat2pmat命令生成pmat文件。接着再使用所述软件工具中的pmat-merge命令对全基因组所有串联的c到t突变信号进行扫描整理并记录成mpmat格式文件。最后使用所述软件工具中的mpmat-select命令进行筛选,获得初步的本发明测序信号。
[0390]
3.本发明富集信号的鉴定
[0391]
在获得初步的本发明测序信号后,需对这些候选区域进行富集检测。首先使用所述软件工具中的find-significant-mpmat命令,对候选区域进行统计检验,统计检验的结果会经过bh方法进行矫正得到假发现率(fdr)。最终认为fdr小于0.01,处理组相比对对照组归一化后的富集倍数大于2,对照组样本中带有突变信号的读段小于3,处理组样本中带有突变信号测序读段不小于5的区域为本发明最终鉴定区域。
[0392]
4.脱靶位点基因序列与sgrna序列的比对
[0393]
在上述步骤鉴定到的富集信号区域中,可以通过序列比对的方法推测sgrna的结合位置。推测出的sgrna结合位置被称为prbs(putative sgrna binding site)。在进行sgrna与富集信号区域内进行序列比对时,使用了改进后的半-全局比对(semi-global alignment)的方法。首先在富集区域内搜索pam序列(nag/ngg),随后对于找到的pam位置,会提取pam 5’方向30nt的序列与sgrna进行半-全局双序列比对,比对中报告的最优结果即为prbs;若在区域内未发现pam,则直接将sgrna与该区域序列进行半-全局比对,比对最优结果为该sgrna的prbs。该步骤使用的比对参数为匹配+5;不匹配-4;打开间隔-24;间隔延伸-8。此步骤的比对程序包含在detect-seq软件工具箱中的mpmat-to-art命令。
[0394]
实验结果:
[0395]
1.含di的模式序列的特异性标记与富集
[0396]
为了证明本发明的方法的特异性和效率,将含有不同修饰碱基的模式序列和对照序列(seq id nos:1,28-30)掺入至建库样品中。最后通过qpcr技术计算和比较了pull-down前后样品中不同模式序列的比例变化(均与不含任何修饰的对照序列(seq id no:1所示的control模式序列)进行相对定量),并计算pull-down前后样品中不同模式序列的富集
倍数。富集倍数如图16所示,由图可知,对于含有单个di:dc和di:dt碱基对的模式序列,本发明的方法可以将之分别富集约220倍和约50倍以上,而只含nick的模式序列几乎完全没有被富集,由此可以证明本发明的方法可以特异且高效地富集含di的dna片段。
[0397]
2.含abe实际编辑位点的dna的富集
[0398]
提取经abemax转染过的hek293t细胞基因组dna,abemax转染细胞的方法参见(xiao wang,et al.nature biotechnology 36,946-949,doi:10.1038/nbt.4198(2018)),通过使用本发明的方法构建出二代测序文库,再经过配套的一系列生物信息学分析,即可获得abemax在全基因组水平编辑位点的信息。图17示出了abe在hek293_site_4(简称为hek4)(seq id no:24)靶向位点(on-target)处的高通量测序结果,由图可知,负对照vector样品没有检测到突变信号,而实验组样品all-pd中有a-to-g的突变信号,其中突变的位置即编辑位点;而且相对于vector样品,all-pd样品中含突变的reads数明显增多,也说明此处确实发生了富集。
[0399]
图18示出了其中一个脱靶位点的高通量测序结果,从图中可以看到vector样品中没有突变信号,而all-pd样品中含有a-to-g的突变信息,也就是脱靶信号。
[0400]
3.本发明的方法检测到的脱靶位点的验证结果
[0401]
图19示出了通过定点深度测序对本发明的方法检测到的其中一个脱靶位点的验证结果,由图可知,该位点的脱靶编辑率高达10.82%。并且从图中on-target序列与此处off-target序列的对比情况可以看到,两者十分接近,推测此处脱靶为cas依赖型脱靶。
[0402]
4.多种abe系统的脱靶效应的评估
[0403]
除abemax系统以外,abe8e和acbe这两种新型工具以及后续可能发展出来的基于腺嘌呤脱氨酶的其他碱基编辑系统,都可以用本发明鉴定脱靶位点。
[0404]
图20-22是将本发明的方法应用到abe8e(richter et al.,2020)和acbe(grunewald et al.,2020;li et al.,2020;sakata et al.,2020;zhang et al.,2020)两种新型工具的脱靶检测时,检测到的on-target和脱靶位点处的高通量测序结果图。对于on-target位点来说,从图20可以观察到这三个系统在sgrna结合区域内部都有对应的a-to-g的突变信号,其中abe8e的信号比abe更强,acbe中除了a-to-g的突变信号外,也有c-to-t的突变信号。
[0405]
对于脱靶位点来说,比如上述提到的off-target 4位点在这三个系统里也都有检测到脱靶信号,只是信号强度不同(图21)。而除了三个系统共有的脱靶位点外,本发明也检测到了abe8e独有的脱靶位点。如图22所示,该位置仅在abe8e系统转染的样品中检测到了脱靶信号,而其他两个样品中并没有检测到相应的脱靶信号。此前文献报道abe8e的活性比abe高得多,而本发明检测到abe8e的脱靶信号也确实多得多,一定程度说明了本发明的可靠性。
[0406]
实施例3
[0407]
本技术发明人将实施例1实验方法步骤7(丙二腈标记步骤)替换为其他5fc标记法后,同样可以促使d5fc处产生c to t突变信号,且不影响富集结果,最终也能实现du位置的标记。
[0408]
以吡啶硼烷等化学标记方法为例,发明人将实施例1中的丙二腈替换为吡啶硼烷(pyridine borane)或2-甲基吡啶硼烷(2-picoline borane)进行反应后(其他实验步骤参
见实施例1),经本发明的方法处理后的spike in模式序列的表征结果如图23所示。图23显示:1)含有单个du:da(seq id no:2)和du:dg(seq id no:5)碱基对的模式序列分别富集了约60倍与20倍,而对于含有ap位点的模式序列(seq id no:4)则几乎完全没有富集(图23a);2)通过sanger测序结果,在含du的模式序列上观察到了连续性的c-to-t突变信号(图23b)。以上结果表明,本发明换用其他类似的化学反应也可引入连续性c-to-t突变信号,且不影响富集结果,最终也能实现du位置的标记。需要指出的是,相比丙二腈标记方法,使用吡啶硼烷标记方法产生的c-to-t突变信号比例较低(图23b)。
[0409]
实施例4
[0410]
实施例1和2中的biotin-du标记分子亦可替换为其他具有富集效果的标记分子,例如,本技术发明人将实施例1中的biotin-du替换为biotin-dg后,含有单个du:da(seq id no:3)和du:dg(seq id no:5)碱基对的模式序列也分别富集了约30倍与20倍,而对于含有ap位点(seq id no:4)、nick(seq id no:30)的模式序列则几乎完全没有富集(图24)。此结果说明换用biotin-dg后,本发明也会特异性地富集含du的dna片段。
[0411]
尽管本发明的具体实施方式已经得到详细的描述,但本领域技术人员将理解:根据已经公开的所有教导,可以对细节进行各种修改和变动,并且这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1