一种N-磺酰脒聚合物的制备方法及应用
一种n-磺酰脒聚合物的制备方法及应用
技术领域
1.本发明属于高分子聚合物合成技术及聚合物催化领域,涉及一种双磺酰叠氮、双醛、环状氨基酸的三组分聚合构建聚(n-磺酰脒)的方法,所制备的聚合物与金属配位后能够作为一种纳米催化剂用于小分子转化反应。
背景技术:
2.具有特定金属配位位点的聚合物,被称为大分子配体,是构建纳米催化剂的优良前体,在纳米催化领域受到了广泛的关注。近些年利用大分子配体与不同金属组合构建人工纳米催化剂的技术层出不穷。同时这人工纳米催化剂可广泛应用于多数小分子反应的转化,有些甚至在细胞质或亚细胞器也可以实现有效催化。
3.大多数大分子配体都是通过引入官能团作为金属配位位点的,这种后修饰策略总是受到侧链修饰效率和多个复杂步骤的限制。目前大多数大分子配体是基于β-酮酯、α-氨基酸功能化的聚合物,如脯氨酸和天冬氨酸,还有一些含氮、膦、硫原子的聚合物。尽管脒作为一种典型的具有过渡金属配位能力的含氮结构,但聚(n-磺酰脒)很少被用于大分子配体的设计。
4.n-磺酰脒类化合物是一种富含氮原子且能与金属强配位的功能结构单元,但是常见的合成的方法存在反应条件苛刻,操作繁琐等缺点,因此也限制了磺酰脒类聚合物的制备。
技术实现要素:
5.为了解决以上技术现有的不足之处与缺点,本发明提供一种基于双磺酰叠氮、双醛、环状氨基酸的三组分聚合制备两性离子和两亲性聚(n-磺酰脒)的方法。该方法具有反应条件温和,所得聚合物分子量高,结构明确的优势。同时以该方法所得到的聚合物可以作为大分子配体,与相应的金属配位后可以作为大分子催化剂有效的催化一系列小分子转化反应,包括水相中通过自组装策略制备纳米级催化剂,可实现催化剂含量在ppm级别的催化反应。
6.本发明的技术方案:
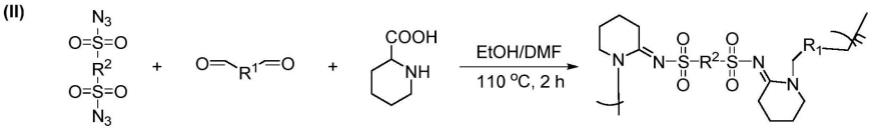
7.一种n-磺酰脒聚合物的制备方法,包括以下制备步骤:
8.准确称量1摩尔当量双磺酰叠氮类单体s-az、1-2摩尔当量双醛类单体ad、1-10摩尔当量的环状氨基酸类单体pro或pip,溶解于溶剂中,得到反应溶液;最终双磺酰叠氮类单体s-az的反应浓度控制在0.025-0.2m;将反应瓶在空气环境中、110-150℃下持续搅拌2-16h,停止反应;使用乙醚和甲醇分别沉降和洗涤三次,离心收集沉淀,产物真空干燥至恒重;
9.反应通式如下:
[0010][0011]
其中,r1为不同长度的醚基或烷基,r2为苯基或苯基衍生物;聚合度n大于5;n-磺酰脒聚合物的重均分子量范围为8000-100000g/mol,分子量分布范围1.1-2.0;
[0012]
所述的双磺酰叠氮类单体s-az是芳香性的双磺酰叠氮;
[0013]
所述的双醛类单体ad选自于不同柔性的双醛;
[0014]
所述的环状氨基酸类单体pro或pip为含氮原子的五元环脯氨酸或六元环哌啶甲酸;
[0015]
所述的溶剂为乙醇、乙腈、四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜中的一种或两种以上以任意比例混合。
[0016]
所述的双磺酰叠氮类单体s-az的结构为:
[0017][0018]
所述的双醛类单体ad的结构为:
[0019][0020]
所述的环状氨基酸类单体pro或pip的结构为:
[0021][0022]
所述的溶剂为体积比为1:1的乙醇与n,n-二甲基甲酰胺的混合溶剂。
[0023]
上述制备方法得到的n-磺酰脒聚合物用于制备纳米粒的应用,步骤如下:将铜催化剂与n-磺酰脒聚合物混合后溶解溶剂中,其中,铜催化剂与磺酰脒官能团的摩尔比为1:1-1:100;在23℃~50℃内搅拌1-10小时成母液,母液中一价铜的浓度为0.1mm-1m;将上述母液加入去离子水中稀释制备粒径范围为10nm-500nm的纳米粒,稀释倍数为1-1000倍,稀
释后的水溶液中的纳米粒作为大分子催化剂用于后续痕量催化反应。
[0024]
所述的溶剂为n,n-二甲基甲酰胺、四氢呋喃或二甲基亚砜。
[0025]
所述的铜催化剂为碘化亚铜、氯化亚铜、溴化亚铜、六氟磷酸四乙腈铜、四氟硼酸四乙腈铜、三氟甲磺酸甲苯铜。
[0026]
上述纳米粒用于催化点击反应,步骤如下:
[0027]
取上述制备好的含有纳米粒的水溶液进一步稀释1-1000倍,向其中加入1摩尔当量的叠氮和1-5摩尔当量的炔烃,使得纳米粒中的一价铜与叠氮的摩尔比为50-10000ppm;反应体系在25-80℃下搅拌1-24小时;反应结束后,通过过滤直接得到产物或以氘代试剂萃取两次,除水,测试核磁氢谱,以核磁氢谱确定转化率。
[0028]
本发明的有益效果:
[0029]
(1)本发明提出了一种基于双磺酰叠氮、双醛、环状氨基酸的三组分聚合一步制备含有磺酰脒结构的三组分聚合物。
[0030]
(2)该类聚合物具有独特的两性离子性质,分子量高(重均分子量范围为8000-100000g/mol)。
[0031]
(3)本发明所提出的合成方法原料易得、反应条件温和、操作步骤简单、副产物少。
[0032]
(4)本发明所得到的聚合物可以作为大分子配体与多种金属配位制备大分子催化剂,可在一价铜与底物摩尔比低于50ppm的剂量下实现催化反应。
[0033]
(5)该聚合物中的磺酰脒类结构片段可以很好的稳定一价铜的价态,在水溶液和空气中可稳定保存。
附图说明
[0034]
图1为本发明实施例1中制备的三组分聚(n-磺酰脒)的核磁图。
[0035]
图2为本发明实施例2中制备的三组分聚(n-磺酰脒)的核磁图。
[0036]
图3为本发明实施例3中制备的三组分聚(n-磺酰脒)的核磁图。
[0037]
图4为本发明实施例4中制备的三组分聚(n-磺酰脒)的核磁图。
[0038]
图5为本发明实施例5中制备的三组分聚(n-磺酰脒)的核磁图。
[0039]
图6为本发明实施例6中制备的三组分聚(n-磺酰脒)的核磁图。
[0040]
图7为本发明实施例7中制备的三组分聚(n-磺酰脒)的核磁图。
[0041]
图8为本发明实施例8中制备的三组分聚(n-磺酰脒)的核磁图。
[0042]
图9为本发明实施例9中制备的三组分聚(n-磺酰脒)的核磁图。
[0043]
图10为本发明实施例10中制备的三组分聚(n-磺酰脒)的核磁图。
[0044]
图11为本发明实施例11中制备的三组分聚(n-磺酰脒)的核磁图。
[0045]
图12为本发明实施例12中制备的三组分聚(n-磺酰脒)的核磁图。
[0046]
图13为本发明实施例13中制备的三组分聚(n-磺酰脒)的核磁图。
[0047]
图14为本发明实施例14中制备的三组分聚(n-磺酰脒)的核磁图。
[0048]
图15为本发明实施例15中制备的三组分聚(n-磺酰脒)的核磁图。
[0049]
图16为本发明实施例16中制备的三组分聚(n-磺酰脒)的核磁图。
[0050]
图17为本发明实施例1中制备的三组分聚(n-磺酰脒)的粒径(a)和电位图(b)。
[0051]
图18为本发明实施例2中制备的三组分聚(n-磺酰脒)的粒径(a)和电位图(b)。
[0052]
图19为本发明实施例3中铜与p1(s-az1/ad1/pro)制备的纳米催化剂的催化效果。
[0053]
图20为本发明实施例4中铜与p1(s-az1/ad1/pro)制备的催化剂72小时的催化效果图。
具体实施方式
[0054]
以下结合附图和技术方案,进一步说明本发明的具体实施方式。
[0055]
实施例1
[0056]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad1(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p1(s-az1/ad1/pro),产率85%,分子量90000g/mol。
[0057]
实施例2
[0058]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad2(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙腈与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p2(s-az1/ad2/pro),产率80%,分子量10000g/mol。
[0059]
实施例3
[0060]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad3(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于四氢呋喃与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在120℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p3(s-az1/ad3/pro),产率85%,分子量9000g/mol。
[0061]
实施例4
[0062]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad4(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与二甲基亚砜的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p4(s-az1/ad4/pro),产率70%,分子量10000g/mol。
[0063]
实施例5
[0064]
在空气环境中将双磺酰叠氮s-az2(0.1mmol,38.0mg)、双醛ad1(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p5(s-az2/ad1/pro),产率70%,分子量16000g/mol。
[0065]
实施例6
[0066]
在空气环境中将双磺酰叠氮s-az2(0.1mmol,38.0mg)、双醛ad2(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙腈与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应
在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p6(s-az2/ad2/pro),产率75%,分子量14000g/mol。
[0067]
实施例7
[0068]
在空气环境中将双磺酰叠氮s-az2(0.1mmol,38.0mg)、双醛ad3(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与二甲基亚砜的混合溶剂(2ml)中,反应在130℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p7(s-az2/ad3/pro),产率70%,分子量13000g/mol。
[0069]
实施例8
[0070]
在空气环境中将双磺酰叠氮s-az2(0.1mmol,38.0mg)、双醛ad4(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于四氢呋喃与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p8(s-az2/ad4/pro),产率80%,分子量70000g/mol。
[0071]
实施例9
[0072]
在空气环境中将双磺酰叠氮s-az3(0.1mmol,38.0mg)、双醛ad1(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在115℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p9(s-az3/ad1/pro),产率70%,分子量10000g/mol。
[0073]
实施例10
[0074]
在空气环境中将双磺酰叠氮s-az3(0.1mmol,38.0mg)、双醛ad2(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙腈与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p10(s-az3/ad2/pro),产率75%,分子量8000g/mol。
[0075]
实施例11
[0076]
在空气环境中将双磺酰叠氮s-az3(0.1mmol,38.0mg)、双醛ad3(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与二甲基亚砜的混合溶剂(2ml)中,反应在130℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p11(s-az3/ad3/pro),产率70%,分子量19000g/mol。
[0077]
实施例12
[0078]
在空气环境中将双磺酰叠氮s-az3(0.1mmol,38.0mg)、双醛ad4(0.1mmol,40.2mg)和脯氨酸pro(0.4mmol,46.0mg)溶解于乙醇与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在110℃下持续2h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p12(s-az3/ad4/pro),产率80%,分子量10000g/mol。
[0079]
实施例13
[0080]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad1(0.1mmol,40.2mg)和哌啶甲酸pip(0.4mmol,58.2mg)溶解于乙醇与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在150℃下持续6h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p13(s-az1/ad1/pip),产率70%,分子量12000g/mol。
[0081]
实施例14
[0082]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad2(0.1mmol,40.2mg)和哌啶甲酸pip(0.4mmol,58.2mg)溶解于乙腈与n,n-二甲基甲酰胺的混合溶剂(2ml)中,反应在150℃下持续6h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p14(s-az1/ad2/pip),产率70%,分子量10000g/mol。
[0083]
实施例15
[0084]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad3(0.1mmol,40.2mg)和哌啶甲酸pip(0.4mmol,58.2mg)溶解于乙醇与二甲基亚砜的混合溶剂(2ml)中,反应在150℃下持续6h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p15(s-az1/ad3/pip),产率75%,分子量12000g/mol。
[0085]
实施例16
[0086]
在空气环境中将双磺酰叠氮s-az1(0.1mmol,38.0mg)、双醛ad4(0.1mmol,40.2mg)和哌啶甲酸pip(0.4mmol,58.2mg)溶解于乙腈与二甲基亚砜的混合溶剂(2ml)中,反应在150℃下持续6h,停止反应冷却至室温。使用乙醚沉降和甲醇洗涤三次,离心收集沉淀,将产物置于真空干燥箱中干燥至恒重得到粉末状产物p16(s-az1/ad4/pip),产率80%,分子量13000g/mol。
[0087]
实施例17
[0088]
称取六氟磷酸四乙腈铜(0.001mmol,0.38mg)与p1(s-az1/ad1/pro)(0.025mmol,21mg)溶于n,n-二甲基甲酰胺(500μl)中,室温下搅拌1小时使得金属与聚合物配位。将溶液滴入10ml纯水中得到具有纳米催化剂的水溶液。取50μl水溶液稀释到500μl作为反应溶剂,向其中加入苯乙炔(0.12mmol,13μl)和苄基叠氮(0.1mmol,12μl)50℃下搅拌12小时(此时铜催化量相对于底物为50ppm)。反应结束后,以氘代氯仿萃取两次,无水硫酸钠干燥,测试核磁氢谱,以核磁氢谱确定转化率。
[0089]
实施例18
[0090]
称取六氟磷酸四乙腈铜(0.001mmol,0.38mg)与p1(s-az1/ad1/pro)(0.025mmol,21mg)溶于n,n-二甲基甲酰胺(500μl)中,室温下搅拌1小时使得金属与聚合物配位,将溶液滴入10ml纯水中得到具有纳米催化剂的水溶液。为了测定一价铜的稳定性,将该水溶液敞口放置在空气中备用。在72h的时候取50μl水溶液稀释到500μl作为反应溶剂(此时铜催化量相对于底物为50ppm)。向其中加入苯乙炔(0.12mmol,13μl)和苄基叠氮(0.1mmol,12μl)50℃下搅拌12小时。反应结束后,以氘代氯仿萃取两次,无水硫酸钠干燥,测试核磁氢谱,以核磁氢谱确定转化率。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1