抑制HDAC6酶的苯并噻二嗪1,1-二氧化物类化合物及其制备方法和应用与流程
本发明涉及药物,尤其涉及抑制hdac6酶的苯并噻二嗪1,1-二氧化物类化合物及其制备方法和应用。
背景技术:
1、组蛋白去乙酰化酶(histone deacetylases,hdacs)和组蛋白乙酰转移酶(histone acetyltransferases,hats)共同调节细胞内乙酰化水平,从而调节基因的表达。hdacs是基因表达的关键调节因子,迄今为止有18hdac亚型在哺乳动物中被鉴定,基于他们对酵母蛋白的同源性分为四类:i类(hdac1、hdac2、hdac3、hdac8)通常存在于细胞核中,并且在各种细胞系和组织中具有普遍存在的表达;ii类进一步分为iia(hdac4、hdac5、hdac7、hdac9)和iib(hdac6、hdac10)两个亚家族对酵母蛋白具有同源性,主要在细胞核和细胞质之间的穿梭;iv类(hdac11)作为其唯一成员,存在细胞核和细胞质中,与i类和ii类酶的催化部位相似。这三类代表zn2+依赖性脱乙酰酶。iii类(sirt1~sirt7)nad+-依赖脱乙酰酶,需要nad+的酵母蛋白sir2的活性和同源物。
2、目前已上市的组蛋白去乙酰化酶抑制剂(hdaci)共有5个,分别为伏立诺他(vorinostat)、贝利司他(belinostat)、帕比司他(panobinostat)、罗米地辛(romidepsin)和西达本胺(chidamide),前三者均为广谱型抑制剂,后两者选择性作用于i类亚型。伏立诺他和罗米地辛用于治疗皮肤t细胞淋巴瘤(ctcl),贝利司他和西达苯胺用于治疗复发及难治性外周t细胞淋巴瘤(ptcl),帕比司他与硼替佐米和地塞米松联用治疗多发性骨髓瘤(mm)。
3、尽管上述hdac抑制剂在临床上已取得良好疗效,但广谱hdac抑制剂普遍存在如下缺点:
4、(1)较强的毒副作用,如恶心、呕吐、骨髓抑制等;
5、(2)基因毒性;
6、(3)药代动力学特性差,生物利用度低、半衰期短等。
7、以上缺点既为肿瘤患者造成不便,也阻碍广谱hdac抑制剂在肿瘤治疗以外领域的应用。
8、目前hdacs亚型选择性抑制剂成为该领域的研究热点,hdac6因其独特的结构和功能,成为肿瘤治疗的新热点。
9、hdac6一种广泛表达的细胞质蛋白去乙酰化酶,主要靶点包括a-微管蛋白和hsp90。通过对这些底物和其他细胞质靶点的翻译后修饰,参与几个关键的细胞过程有关,包括原代细胞纤毛、细胞内信号传导和dna损伤反应,抑制hdac6会导致细胞纤毛的恢复和恶性表型的衰减,与它在细胞调节中的作用一致。同时抑制hdac6已被证明可以减少致癌的hedgehog信号通路(hedgehog信号通路控制细胞命运、增殖与分化,该信号通路被异常激活时,会引起肿瘤的发生与发展。)hdac6通过与信号中介体的相互作用或通过调节hsp90直接地与细胞内信号传导,是一个重要的细胞内伴侣。总的来说,这些研究将hdac6与多种致癌过程联系起来,并强调了hdac6抑制剂诱导细胞和免疫介导的抗肿瘤活性的潜力。
10、hdac6众所周知的底物之一是α-微管蛋白,它是微管的主要成分。α-微管蛋白在40位赖氨酸处乙酰化是微管中的一个常见过程。微管功能的稳定性强烈依赖于α-微管蛋白的乙酰化状态。基于微管的转运障碍可能会扰乱神经元细胞体和轴突/树突之间的线粒体转运,并进一步导致线粒体功能障碍和随后的细胞死亡。线粒体是突出的基于微管的轴突转运细胞器,通过抑制hdac6提高α-微管蛋白的乙酰化可以改善基于微管的运输,从而改善线粒体的转运缺陷。现有研究显示,hdac6抑制可能会减缓或逆转与aβ相关的神经元损伤,因此代表了治疗ad的可行药物靶点。
技术实现思路
1、本发明提供了一种结构新颖的苯并噻二嗪1,1-二氧化物类化合物,可选择性抑制hdac6酶,对神经细胞具有较强的保护作用,毒性低、潜在心脏毒性小,有望作为神经保护剂用于神经退行性疾病的治疗。
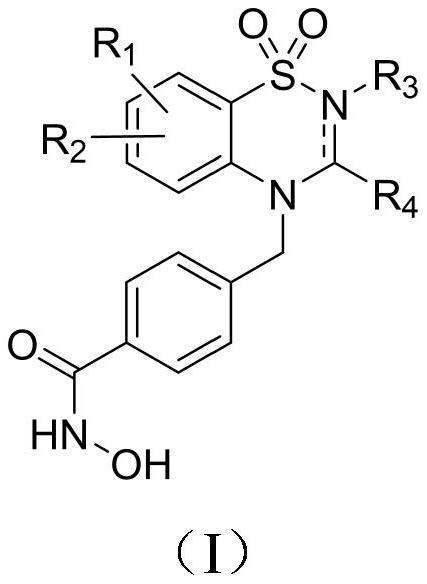
2、为了实现上述发明目的,本发明的第一方面提供了一种苯并噻二嗪1,1-二氧化物类化合物,结构通式如式(ⅰ)所示,或其异构体,或其药学上可接受的盐、酯或前药;
3、
4、其中,
5、r1和r2独立地选自氢、氘、羟基、卤素、烷基、烷氧基、环烷基、苄基、杂环烷基、芳基、杂芳基、氰基、卤代烷烃、酰基、磺酰基或氨基烷基,其可任选地被取代;
6、当1,1-二氧基苯并噻二嗪环上的2-n与3-c之间为n=c双键时,式(ⅰ)化合物不包含r3;
7、当1,1-二氧基苯并噻二嗪环上的2-n与3-c之间为n-c单键时,r3选自氢、烷基、环烷基、芳基、杂芳基、杂环烷基、酰基、磺酰基或-(chm)n-r5,其可任选地被取代;r5选自环烷基、芳基、杂芳基或羟基;m为0~2的整数,n为正整数;
8、r4选自氢、羟基、氧基、烷基、环烷基、苄基、芳基、杂芳基、杂环烷基、酰基或磺酰基,其可任选地被取代。
9、本发明优选的,苯并噻二嗪1,1-二氧化物类化合物包括化合物(ⅰ-1)或化合物(ⅰ-2):
10、
11、在式(ⅰ-1)化合物中,
12、r1和r2独立地选自氢、氘、羟基、卤素、烷基、烷氧基、环烷基、苄基、杂环烷基、芳基、杂芳基、氰基、卤代烷烃、酰基、磺酰基或氨基烷基,其可任选地被取代;
13、r4选自氢、羟基、氧基、烷基、环烷基、苄基、芳基、杂芳基、杂环烷基、酰基或磺酰基,其可任选地被取代;
14、
15、在式(ⅰ-2)化合物中,
16、r1和r2独立地选自氢、氘、羟基、卤素、烷基、烷氧基、环烷基、苄基、杂环烷基、芳基、杂芳基、氰基、卤代烷烃、酰基、磺酰基或氨基烷基,其可任选地被取代;
17、r3选自氢、烷基、环烷基、芳基、杂芳基、杂环烷基、酰基、磺酰基或-(chm)n-r5,其可任选地被取代;r5选自环烷基、芳基、杂芳基或羟基;m为0~2的整数,n为正整数;
18、r4选自氢、羟基、氧基、烷基、环烷基、苄基、芳基、杂芳基、杂环烷基、酰基或磺酰基,其可任选地被取代。
19、本发明优选的,所述的烷基为含有1~4个碳原子的烷基,其可任选地被0~3个卤素取代;
20、本发明优选的,所述的环烷基为含有3~6个碳原子的环烷基,其可任选地被0~3个卤素取代;
21、本发明优选的,所述的杂环烷基选自吡咯基、吗啉基、哌啶基、哌嗪环、四氢喹啉基、四氢三唑并吡嗪基、二氮杂环庚烷基或哌嗪基,其可任选地被取代;
22、本发明优选的,所述的芳基或杂芳基选自苯基、萘基、蒽基、吡啶基、嘧啶基、吡嗪基、吲哚基、咪唑基、苯并噁唑基、苯并呋喃基、苯并噻吩基、苯并噻唑基、三唑基、异噁唑基、喹啉基、吡咯基、吡唑基或5,6,7,8-四氢异喹啉;其可任选地被取代;
23、本发明优选的,所述的酰基选自乙酰基、丙酰基、异丁酰基或芳基酰基,其可任选地被取代;
24、本发明优选的,所述的磺酰基选自甲磺酰基或芳基磺酰基,其可任选地被取代;
25、本发明优选的,所述的氨基烷基选自二甲氨基烷基、甲基氨基烷基、哌嗪烷基或哌啶烷基,其可任选地被取代;
26、本发明优选的,所述的卤素选自氟或氯;
27、本发明优选的,所述的烷氧基选自含有1~4个碳原子的烷氧基;
28、本发明优选的,所述的r5选自芳基、环烷基或羟基;
29、本发明优选的,所述的n选自0~4的整数。
30、本发明优选的,式(ⅰ)化合物药学上可接受的盐包括式(ⅰ)化合物与盐酸、氢溴酸、硫酸、醋酸、三氟醋酸、柠檬酸、酒石酸、马来酸、富马酸、甲磺酸、苹果酸、对甲苯磺酸或草酸反应生成的阴离子盐;或式(ⅰ)化合物与钠离子溶液、钾离子溶液反应生成的阳离子盐。
31、本发明进一步优选的,r1和r2独立地选自氢、甲基、f或cl;
32、本发明进一步优选的,r3连接的n原子与相邻c原子为n=c双键,式(ⅰ)化合物不包含r3;或,r3连接的n原子与相邻c原子为n-c单键,r3选自氢或-(chm)n-r5,r5选自苯基、环丙烷基、环己烷基或羟基;m为1或2,n为1、2或3;
33、本发明进一步优选的,r4选自氢、氧基或甲基。
34、在本发明的一些具体实施方式中,苯并噻二嗪1,1-二氧化物类化合物包括表1所示的化合物或其异构体,或其药学上可接受的盐、酯或前药:
35、表1
36、
37、
38、
39、
40、
41、
42、本发明的第二方面提供了一种上述技术方案所述苯并噻二嗪1,1-二氧化物类化合物的制备方法,包括下述方法中的一种或多种:
43、若式(ⅰ)化合物不包含r3,采用方法a制备;
44、方法a包括以下步骤:
45、a1,式(ⅱ)化合物与4-溴甲基苯甲酸甲酯在加热条件下反应,得到式(ⅲ)化合物;
46、
47、其中,r1、r2、r4如上述技术方案所示;
48、本发明优选的,步骤a1所述的加热温度为60℃~80℃,加热反应时间为3~4h;
49、a2,式(ⅲ)化合物与羟胺在碱性条件下反应,得到式(ⅰ-1)化合物;
50、
51、其中,r1、r2、r4如上述技术方案所示;
52、本发明优选的,步骤a2中,式(ⅲ)化合物与羟胺在甲醇钠甲醇溶液中进行反应;
53、本发明优选的,步骤a2中,反应温度为-5℃~5℃,反应温度为4~6h;
54、若式(ⅰ)化合物包含r3且r3为氢、r4为非氧基,可采用方法b制备:
55、方法b包括以下步骤:
56、b1,式(ⅲ)化合物经硼氢化钠还原为式(ⅳ)化合物;
57、
58、其中,r1、r2如上述技术方案所示;r4选自上述技术方案所示的非氧基基团;
59、本发明优选的,步骤b1中,将氢化钠加入式(ⅲ)化合物的溶液中进行还原反应,生成式(ⅳ)化合物;
60、b2,式(ⅳ)化合物与羟胺在碱性条件下反应,得到式(ⅰ-2-1)化合物;
61、
62、其中,r1、r2如上述技术方案所示;r4选自上述技术方案所示的非氧基基团;
63、本发明优选的,步骤b2中,式(ⅳ)化合物与羟胺在甲醇钠甲醇溶液中进行反应;
64、本发明优选的,步骤b2中,反应温度为-5℃~5℃,反应温度为4~6h;
65、若式(ⅰ)化合物包含r3且r3为氢、r4为氧基,采用方法c制备:
66、方法c包括以下步骤:
67、c1,式(ⅴ)化合物与4-溴甲基苯甲酸甲酯在碱性条件下反应得到式(ⅵ)化合物;
68、
69、其中,r1、r2如上述技术方案所示;
70、本发明优选的,步骤c1中,式(ⅴ)化合物、碳酸钠溶解于n,n-二甲基甲酰胺中,将4-溴甲基苯甲酸甲酯逐滴加入上述溶液中,反应得到式(ⅵ)化合物;
71、本发明优选的,步骤c1中,反应时间为3~5h;
72、c2,式(ⅵ)化合物与羟胺在碱性条件下反应得到式(ⅰ-2-2)化合物;
73、
74、其中,r1、r2如上述技术方案所示;
75、若式(ⅰ)化合物包含r3且r3为非氢,采用方法d制备:
76、本发明优选的,步骤c2中,式(ⅵ)化合物与羟胺在甲醇钠甲醇溶液中进行反应;
77、本发明优选的,步骤c2中,反应温度为-5℃~5℃,反应温度为4~6h;
78、方法d包括以下步骤:
79、d1,式(ⅳ)化合物与式(ⅶ)化合物在加热条件下反应得到式(ⅷ)化合物;
80、
81、其中,
82、式(ⅳ)化合物可按照步骤b1所示方法制备得到;
83、r1、r2、r4如上述技术方案所示;r3选自上述技术方案所示的非氢基团;
84、x为卤素;
85、本发明优选的,步骤d1所述的加热温度为60℃~80℃,反应时间为1.5~3h;
86、d2,式(ⅷ)化合物与羟胺在碱性条件下反应得到式(ⅰ-2-3)化合物;
87、
88、其中,
89、r1、r2、r4如上述技术方案所示;r3选自上述技术方案所示的非氢基团。
90、本发明优选的,步骤d2中,式(ⅷ)化合物与羟胺在甲醇钠甲醇溶液中进行反应;
91、本发明优选的,步骤d2中,反应温度为-5℃~5℃,反应温度为4~6h。
92、本发明优选的,所述的式(ⅴ)化合物制备方法包括以下步骤:
93、式(ⅸ)化合物在-50℃~100℃与氯磺酰异氰酸酯反应后,再在30℃~120℃下与氯化铝反应,得到式(ⅴ)化合物;
94、
95、其中,r1、r2如上述技术方案所示;
96、本发明进一步优选的,式(ⅴ)化合物制备方法中,式(ⅸ)化合物与氯磺酰异氰酸酯的反应时间为20~60min;
97、本发明进一步优选的,式(ⅴ)化合物制备方法中,中间体1与氯化铝的反应时间为0.5~2h;
98、本发明优选的,所述的式(ⅱ)化合物制备方法包括以下步骤:
99、e1,式(ⅴ)化合物与稀硫酸在130℃~150℃下反应,得到式(ⅹ)化合物;
100、
101、其中,r1、r2如上述技术方案所示;
102、本发明进一步优选的,步骤e1所述的稀硫酸为体积浓度45~60%的硫酸水溶液;
103、本发明进一步优选的,步骤e1中,反应时间为6~10h;
104、e2,式(ⅹ)化合物与式(ⅺ)化合物加热反应,得到式(ⅱ)化合物;
105、
106、其中,r1、r2、r4如上述技术方案所示。
107、本发明进一步优选的,步骤e2中,加热温度为90℃~110℃,反应时间为1.5~3h。
108、本发明的第三方面提供了一种用于制备前述技术方案所述苯并噻二嗪1,1-二氧化物类化合物的中间体化合物,包括下述化合物中的一种或多种:
109、式(ⅱ)化合物或其异构体、药学上可接受的盐、酯或前药:
110、
111、其中,r1、r2、r4如前述技术方案所示;
112、和/或,式(ⅲ)化合物或其异构体、药学上可接受的盐、酯或前药:
113、
114、其中,r1、r2、r4如前述技术方案所示;
115、和/或,式(ⅳ)化合物或其异构体、药学上可接受的盐、酯或前药:
116、
117、其中,r1、r2如前述技术方案所示;r4选自前述技术方案所示的非氧基基团;
118、和/或,式(ⅴ)化合物或其异构体、药学上可接受的盐、酯或前药:
119、
120、其中,r1、r2如前述技术方案所示;
121、和/或,式(ⅵ)化合物或其异构体、药学上可接受的盐、酯或前药:
122、
123、其中,r1、r2如前述技术方案所示;
124、和/或,式(ⅷ)化合物或其异构体、药学上可接受的盐、酯或前药:
125、
126、其中,r1、r2、r4如前述技术方案所示;r3选自前述技术方案所示的非氢基团;
127、和/或,式(ⅸ)化合物或其异构体、药学上可接受的盐、酯或前药:
128、
129、
130、其中,r1、r2如前述技术方案所示;
131、和/或,式(ⅹ)化合物或其异构体、药学上可接受的盐、酯或前药:
132、
133、其中,r1、r2如前述技术方案所示。
134、本发明的第四方面提供了前述技术方案所述苯并噻二嗪1,1-二氧化物类化合物、前述技术方案所述制备方法得到的苯并噻二嗪1,1-二氧化物类化合物或上述技术方案所述的中间体化合物在制备组蛋白去乙酰化酶抑制剂或神经退行性疾病治疗药物中的应用。
135、本发明优选的,所述组蛋白去乙酰化酶抑制剂为hdac6和/或hdac1抑制剂。
136、本发明优选的,所述的神经退行性疾病治疗药物包括神经退行性疾病包括急性神经退行性疾病和慢性神经退行性疾病的治疗药物,急性神经退行性疾病主要包括脑缺血(ci)、脑损伤(bi)、癫痫;慢性神经退行性疾病包括阿尔茨海默病(ad)、帕金森病(pd)、亨廷顿病(hd)、肌萎缩性侧索硬化(als)、不同类型脊髓小脑共济失调(sca)、pick病等。
137、本发明的第五方面提供了一种药物组合物,包括至少一种活性组分以及一种或多种药学上可接受的辅料;所述活性组分包括前述技术方案所述苯并噻二嗪1,1-二氧化物类化合物或前述技术方案所述制备方法得到的苯并噻二嗪1,1-二氧化物类化合物。
138、本发明优选的,所述药学上可接受的辅料包括稀释剂、赋形剂、填充剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂、香味剂和甜味剂中的一种或多种。
139、本发明所述药物组合物可以制成片剂,粉剂,粒剂,胶囊,口服液及注射用药等多种形式,上述各剂型的药物均可以按照药学领域的常规方法制备。本发明所述药物组合物中的活性组分还可以与其他具有治疗效果或增强治疗效果、降低毒副作用、延长代谢时间的有效成分共同组成药物组合物。
140、与现有技术相比,本发明的有益效果:
141、1)本发明所述苯并噻二嗪1,1-二氧化物类化合物对hdac6和hdac1均具有抑制活性,对hdac6显示出较强的抑制活性;多数化合物对hdac6抑制活性优于hdac1,显示出一定的选择性(相较于hdac1);
142、2)本发明所述苯并噻二嗪1,1-二氧化物类化合物可提高sh-sy5y细胞损伤模型的细胞活力,显示出较好的细胞损伤保护作用;
143、3)本发明所述苯并噻二嗪1,1-二氧化物类化合物对人神经母细胞瘤细胞(sh-sy5y)、人正常胚肺成纤维细胞(mrc-5)的抗增殖作用普遍较弱,且弱于现有药物sw-100和acy-1215,对细胞毒性更小;
144、3)本发明所述苯并噻二嗪1,1-二氧化物类化合物对herg抑制活性弱,潜在心脏毒性较小。
- 还没有人留言评论。精彩留言会获得点赞!