一种DNA甲基化检测方法及所用试剂
一种dna甲基化检测方法及所用试剂
技术领域
1.本发明涉及生物医学领域,特别是涉及一种dna甲基化检测方法及所用试剂。
背景技术:
2.表观遗传学改变在癌症的发生发展中起着重要的作用。表观遗传学指在dna序列不发生改变的情况 下引起基因表达或功能变化,主要包括dna甲基化、组蛋白修饰等
1.。dna甲基化是最主要的表观遗传 学修饰之一,参与细胞分化和发育、肿瘤发生和其他疾病。在正常哺乳动物细胞中,胞嘧啶甲基化(5-m
c) 几乎仅存在于cpg二核苷酸中。一般情况下,70%-90%的散在cpg位点是甲基化的,而未甲基化的cpg 通常只存在于被称为cpg岛的dna区域。cpg岛是基因组中cpg二核苷酸的百分比高于随机分布的cpg 二核苷酸的预期区域,其所在位置多为基因启动子区域,与人类基因组编码基因相关,正常状态下通常处 于低甲基化状态
2.。然而,对癌症表观基因组的全基因组研究表明,肿瘤细胞基因组中部分cpg岛存在异 常甲基化,表明cpg岛甲基化可能参与肿瘤的发生发展过程
3.。基因启动子区域cpg岛的高甲基化可以 抑制下游基因的表达,促进肿瘤发生。研究表明,异常dna甲基化在癌症中广泛存在,可能是肿瘤发生 过程中最早发生的变化之一
4.。与体细胞突变分析相比,dna甲基化分析在癌症检测方面具有以下优势: (1)异常甲基化在癌症早期就出现;(2)甲基化发生频率较基因突变更为频繁;(3)dna甲基化多为 连续位点发生,有多个可检测的甲基化靶区,以及每个目标基因组区域内多个改变的cpg位点
5.。
3.dna甲基化的检测方法有多种,亚硫酸氢盐处理转化法一直是dna甲基化分析的标准方法。该方法 于1992年由marianne frommer首次提出
6.,其原理是在酸性和高温条件下,亚硫酸氢盐将未甲基 化的胞嘧啶(c)脱氨转化成尿嘧啶(u),通过pcr扩增将尿嘧啶(u)解读为胸腺嘧啶(t),而甲基化 的胞嘧啶(mc)保持不变,随后用测序、定量pcr和芯片技术分析比较转化和未转化的dna序列,来判 断dna甲基化水平。基于亚硫酸氢盐的转化技术已经成为许多技术的基础,如wgbs、rrbs、mctaseq、 靶向亚硫酸氢盐测序、甲基化阵列、msp等
7.。
4.然而,亚硫酸氢盐转化法有两大主要缺点:第一,亚硫酸氢盐处理是一种苛刻的化学反应,在酸性和 高温条件下,由于脱嘧啶作用,大部分dna会被降解,导致该技术需要较多的起始量dna,因此在微量 dna样本中应用严重受限。第二,未修饰的胞嘧啶约占人类基因组中总胞嘧啶的95%,亚硫酸氢盐转化 将未甲基化的胞嘧啶全部转化为尿嘧啶,经pcr扩增被识别为胸腺嘧啶,导致基因组由原本atcg四种 碱基平衡组成降低为仅剩余atg碱基为主,严重降低了dna序列复杂度,导致引物探针设计困难、扩增 偏倚大、测序质量差、基因组覆盖不均和测序成本增加。且由于dna序列复杂度降低,多重pcr扩增设 计尤其困难。
5.使用甲基化敏感的限制性内切酶切割特定核苷酸序列也是dna甲基化研究的经典方法。这里使用的 甲基化敏感限制性内切酶(如hpaii、bstui、acii和hin6i)切割未甲基化区域,当目标序列中的胞嘧啶 残基被甲基化时,这些酶将不能切断识别序列。gabriel beikircher等人将该技术和qpcr技术相结合,开 发了甲基化敏感的限制性内切酶(msre)
profilesenableearlydiagnosis,prognosisprediction,andscreeningforcolorectalcancer.scitranslmed2020,12(524):eaax7533.
15.[6]frommerm,mcdonaldle,millards,etal.agenomicsequencingprotocolthatyieldsapositivedisplayof5-methylcytosineresiduesinindividualdnastrands.procnatlacadsciusa1992,89(5):1827-1831.
[0016]
[7]sharmam,vermark,kumars,etal.computationalchallengesindetectionofcancerusingcell-freednamethylation.computstructbiotechnolj2022,2026-39.
[0017]
[8]beikircherg,pulvererw,hofnerm,etal.multiplexedandsensitivednamethylationtestingusingmethylation-sensitiverestrictionenzymes"msre-qpcr".methodsmolbiol2018,1708407-424.
[0018]
[9]liuy,siejka-zieli
ń
skap,velikovag,etal.bisulfite-freedirectdetectionof5-methylcytosineand5-hydroxymethylcytosineatbaseresolution.natbiotechnol2019,37(4):424-429.
[0019]
[10]skvortsovak,stirzakerc,taberlayp.thednamethylationlandscapeincancer.essaysbiochem2019,63(6):797-811.
[0020]
[11]jasminef,haqz,kamalm,etal.interactionbetweenmicrosatelliteinstability(msi)andtumordnamethylationinthepathogenesisofcolorectalcarcinoma.cancers(basel)2021,13(19):4956.
[0021]
[12]depalmafde,d'argeniov,polj,etal.themolecularhallmarksoftheserratedpathwayincolorectalcancer.cancers(basel)2019,11(7):1017。
技术实现要素:
[0022]
本发明要解决的技术问题是提供一种dna甲基化检测方法、引物探针组合及其应用。
[0023]
为解决上述技术问题,本发明提供一种用于检测甲基化标志物的试剂(即,用于检测甲基化标志物的试剂在制备试剂盒中的用途):所述甲基化标志物为sdc2-1、sdc2-2、sdc2-4、ndrg4-1、ndrg4-2、ndrg4-3、ndrg4-4、ndrg4-5、ndrg4-7中的至少任一。
[0024]
说明:试剂(试剂盒)用于诊断受试者中是否存在结直肠良性息肉、进展性腺瘤或结直肠癌;所述方法包括检测来自所述受试者的组织或粪便样本中的dna中的甲基化标志物以判断所述dna的甲基化水平,如果所述甲基化水平高于正常对照样本的dna甲基化水平,则确定所述受试者中存在结直肠良性息肉、进展性腺瘤或结直肠癌,所述甲基化标志物的检测是使用tap转化的多重定量甲基化特异性pcr进行的。
[0025]
标志物所在染色体位置具体如下:
[0026]
标志物名称标志物所在染色体位置(hg38)sdc2-1chr8:96494038-96494125(+)sdc2-2chr8:96494036-96494125(-)sdc2-4chr8:96494139-96494216(-)ndrg4-1chr16:58463404-58463474(+)
ndrg4-2chr16:58463428-58463505(-)ndrg4-3chr16:58463519-58463586(+)ndrg4-4chr16:58463521-58463587(-)ndrg4-5chr16:58463627-58463690(+)ndrg4-7chr16:58463740-58463826(+)
[0027]
作为本发明的试剂的改进:所述甲基化标志物为sdc2-1、sdc2-2、sdc2-4、ndrg4-1、ndrg4-2、 ndrg4-3、ndrg4-4、ndrg4-5和ndrg4-7的组合。
[0028]
作为本发明的试剂的进一步改进:甲基化标志物的引物和探针为:
[0029][0030][0031]
说明:根据甲基化标志物所在基因及染色体位置进行设计。
[0032]
作为本发明的试剂的改进,试剂盒包括:kapa probe fast qpcr master mix、如
上所述的引物和 探针、内参基因引物混合物、内参基因探针、去酶水、rox。
[0033]
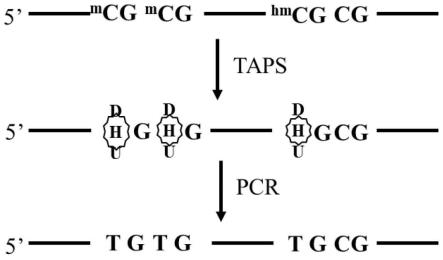
本发明还同时提供了一种dna甲基化检测方法:使用tet酶将mc氧化为fc和cac,经过吡啶硼烷 还原为dhu,而保存未甲基化的c保持不变。
[0034]
说明:所述探针的荧光标记可以为fam,vic,ned,cy5,tet,joe,tamra,等已知能够作为 探针荧光标记的荧光基团。所述的内参基因为actin,内参基因也可以在本专业人员的知识下改为其他在 基因组中拷贝数稳定存在的基因区域。
[0035]
为降低检测成本,提高检测的灵敏性,本发明将taps技术和qpcr技术相结合开发出一种新型的dna 甲基化检测方法。本发明利用taps技术建立的taps-qpcr检测技术,用于结直肠癌组织标本、结直肠 癌患者和健康志愿者的粪便标本的甲基化水平检测,以期将该技术用于结直肠癌的诊断。
[0036]
本发明在发明过程中:
[0037]
通过收集入组80份组织(i、ii、iii、iv期期结直肠癌病人的癌组织各10例,10例进展性腺瘤组织、 10例良性息肉组织,20例癌旁正常组织),将同一类型和分期的组织提取dna并混成dna混合物,进 行亚硫酸氢盐转化和两步pcr建库扩增子测序,根据测序结果验证能清晰区分肿瘤组织和正常组织的 sdc2、ndrg4基因启动子dna甲基化区域。
[0038]
在此基础上,将taps技术和多重荧光定量pcr(qpcr)技术相结合进行dna甲基化水平检测,通 过对单重反应和多重qpcr反应条件进行逐步优化,得到一种灵敏且稳定的dna甲基化多重检测方法, 最后通过检测结直肠癌患者组织dna及粪便dna验证了本发明用于dna甲基化检测的可行性和实用性。
[0039]
本发明利用qpcr技术,设计了定量检测技术,对多个标志物进行了定量测定。首先根据筛选出的多 个结直肠癌特异的甲基化标志物分别设计正链或负链taps转化特异性引物和探针,设计示意图见图2; 然后对单个标志物进行肿瘤特异性测定和多重pcr反应条件逐步优化,得到一种灵敏且稳定的荧光定量检 测方法,可在单管反应中对多个标志物进行同时测定,从而表现多个标志物的总体甲基化水平,低至0.5% 甲基化水平可稳定检测到。
[0040]
采用本发明的方法对多种类型的组织样品,包括结直肠癌、进展性腺瘤、肠息肉、健康对照组织样品 进行检测,同时进行粪便标本检测,证明本方法的可行性。
[0041]
本发明的技术要点是根据taps转化的原理设计taps转化后甲基化特异性的引物和探针,并将其组 合为多重pcr反应并赋予其相同的荧光信号。通过设置内参assay,分析靶标序列与内参assay之间荧光 信号的差值,判断样品的甲基化水平。
[0042]
本发明的技术点在于:
[0043]
本发明利用亚硫酸氢盐扩增子测序技术,验证结直肠癌组织中能够区分肿瘤、息肉和健康对照组织的 特异性dna甲基化基因,并选定候选基因的差异甲基化区域。
[0044]
在此基础上,本发明通过设计taps转化特异性的引物和探针组合,并优化多重扩增反应形成本发明 方案。本发明方案的技术要点是设计的引物和探针仅能扩增被taps技术成功转化的含有甲基化的cg位 点的序列。
[0045]
进一步,通过优化反应条件,将10重pcr合并在同一管中进行反应,并设计内参反应,用于分析甲 基化程度。
[0046]
进一步,通过入组10对crc患者的肿瘤组织及配对的正常组织,10例良性息肉患者和10例进展性 息肉患者,使用本发明的tap-mqpcr(tap based multiplex qpcr)方法对这
些组织样本进行检测,结果 显示该方法所检测的甲基化水平差异能准确区分正常组织、良性息肉、进展性腺瘤组织及肿瘤组织。使用 one-way anova统计学方法分析,良性息肉,进展性腺瘤,肿瘤组织分别与正常组织具有统计学差异。 对正常组织和crc肿瘤组织进行受试者工作特征曲线(receiver operating characteristic curve,roc曲线) 分析,结果显示,该方法对crc肿瘤组织检测能力较强,曲线下面积auc值为0.98,在特异性为100% 情况下,敏感性为90%。
[0047]
本发明方案,无需对样品进行亚硫酸盐转化,克服了传统亚硫酸盐转化导致的dna断裂、序列碱基 不平衡问题,保留了原始dna序列的复杂度,易于设计多重引物和探针。本发明仅扩增被taps转化后 的含有甲基化的cg位点的序列,引物和探针设计时单个引物或探针上至少存在2个taps转化后的cg 位点,且前向和反向引物与探针三者所在序列必须均甲基化且均被taps成功转化才能被扩增,保证了本 方案甲基化检测的高度特异性。
[0048]
本发明将taps技术和qpcr技术相结合研发出一种新型的dna甲基化检测方法,即,是基于taps 技术的qpcr检测技术用于dna甲基化的检测。taps转化技术已经被发明人申请专利(申请号ca3087915, pct/us2019/012627,wo2019136413a1),但是用于qpcr和用于多重qpcr技术目前均没有报道,本发 明是在上述技术基础上的再创新。
[0049]
采用本发明的dna甲基化检测方法,具有如下的技术优势:
[0050]
本发明由于采用taps,因此无需亚硫酸氢盐转化,克服传统亚硫酸盐转化的序列简并性和dna分 子断裂的缺陷,引物设计更简单。
[0051]
本发明根据taps转化的原理,设计了引物3’末端与甲基化cg位点匹配的引物,保证了pcr扩增 的特异性;每组扩增子2条引物均至少满足每条引物上至少2个待测的转化后的cg位点,探针至少覆盖 2个转化后的cg位点,因此严格保障了扩增的甲基化特异性。
[0052]
本发明采用qpcr检测技术,适用于起始量较低的dna,且由于qpcr检测仪器较为普及。因此更 容易推广和应用。
[0053]
本发明通过引物探针的设计,相比于目前的高通量测序检测,方法更简单、更方便、检测周期更短; 设计了多重qpcr方法,因此检测灵敏度更高。
附图说明
[0054]
下面结合附图对本发明的具体实施方式作进一步详细说明。
[0055]
图1为taps技术原理示意图;
[0056]
图2为taps-qpcr设计示意图:a.sdc2多重assay设计示意图;b.ndrg4多重assay设计示意图; c.qpcr引物探针设计原理示意图。阴影区域示甲基化cg位点,经taps转换后形成tg序列。
[0057]
图3为候选基因在不同类型组织样本中的甲基化水平,横轴为候选基因的cg位点,纵轴为甲基化水 平。
[0058]
图4为单个甲基化标志物的扩增曲线图,fam表示甲基化荧光信号,cy5表示内参基因荧光信号。
[0059]
图5为单个甲基化标志物肿瘤特异性测试结果,横轴为每一个标志物,纵轴越小表示该标志物区分肿 瘤组织和正常组织的能力越强。
[0060]
图6为tap-mqpcr灵敏度检测扩增曲线。
[0061]
图7为单一甲基化标志物和多个甲基化标志物联合检测的灵敏度比较。
[0062]
图8为tap-mqpcr检测不同类型组织dna甲基化水平。
[0063]
图9为tap-mqpcr检测肿瘤组织与正常组织的roc分析结果。
[0064]
图10为tap-mqpcr检测健康人及结直肠癌患者粪便dna甲基化水平.
[0065]
图11为tap-mqpcr检测结直肠癌患者粪便dna和健康人粪便dna的roc分析结果。以-11.75 为阈值,在特异性为85.71%时,敏感性为52.94%。
具体实施方式
[0066]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0067]
实施例1:结直肠癌组织特异性差异甲基化基因区域验证
[0068]
利用亚硫酸氢盐扩增子测序技术(参照已公开发表于《development of a dual-index sequencing strategyand curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform》的两 步pcr构建文库的方法),验证结直肠癌组织中能够区分肿瘤、息肉和健康对照组织的特异性dna甲基 化基因(sdc2、ndrg4和bmp3基因),并选定候选基因的差异甲基化区域。
[0069]
所得结果为:sdc2和ndrg4能够明显区分来源于中国人的结直肠癌、息肉组织和癌旁组织;bmp3 的启动子区域dna甲基化不能有效区分来源于中国人的结直肠癌、息肉组织和癌旁组织。如图3所示。
[0070]
以图3a的“sdc2-正链平均甲基化水平”和为以图3b的“sdc2-负链平均甲基化水平”例进行如下 的具体说明:
[0071]
normal代表结直肠癌手术切缘5cm外的正常结直肠组织;poly代表结直肠息肉组织;crc tumor1/2 代表临床分期为i和ii期的结直肠癌患者的手术切除癌组织;crc tumor3/4代表临床分期为iii和iv期 的结直肠癌患者的手术切除癌组织;该数据显示:sdc2基因启动子区域的正链甲基化水平,在不同分期 结直肠癌组织、结直肠息肉组织中的均高于正常结直肠组织;sdc2基因启动子区域的负链甲基化水平, 在不同分期结直肠癌组织、结直肠息肉组织中的均高于正常结直肠组织。
[0072]
因此,根据图3,可得知:结直肠癌组织和息肉组织的sdc2基因和ndrg4基因的启动子区域正、 负链甲基化均高于正常结直肠组织,sdc2和ndrg4基因启动子区域甲基化能够区分异常的结直肠癌组 织、结直肠息肉组织与正常结直肠组织。因此,可以作为结直肠癌组织、结直肠息肉组织与正常结直肠组 织dna甲基化差异的标志物。
[0073]
结直肠癌组织和息肉组织的bmp3基因的启动子区域正、负链甲基化在正常结直肠组织、结直肠癌组 织、结直肠息肉组织中甲基化水平高低不等,bmp3基因启动子区域甲基化不能区分异常的结直肠癌组织、 结直肠息肉组织与正常结直肠组织。
[0074]
实施例2:taps-qpcr单一标志物靶标检测5%甲基化dna样品
[0075]
针对验证后的sdc2、ndrg4启动子区域分别设计taps正、负链转化特异的引物和探针,设计原则 为:引物的3’端包含2个或2个以上的cg位点;探针横跨2个或2个以上的cg位点,保证taps转化 的特异性;引物和探针序列与taps转化后cg转化为tg的序列相同或互补配对。引物、探针的设计原 理和原则见图2。
[0076]
在管家基因actin基因区域设计引物和探针结合区域不含cg位点的扩增子作为内参区域用于质控样 品量,简单评估甲基化水平。
[0077]
甲基化标志物所在染色体位置见表1,设计的引物及探针序列见表2,内参基因的引物和探针序列见 表3。
[0078]
表1.本发明筛选得到的甲基化标志物所在基因及染色体位置
[0079]
标志物名称标志物所在染色体位置(hg38)sdc2-1chr8:96494038-96494125(+)sdc2-2chr8:96494036-96494125(-)sdc2-3chr8:96494169-96494239(+)sdc2-4chr8:96494139-96494216(-)ndrg4-1chr16:58463404-58463474(+)ndrg4-2chr16:58463428-58463505(-)ndrg4-3chr16:58463519-58463586(+)ndrg4-4chr16:58463521-58463587(-)ndrg4-5chr16:58463627-58463690(+)ndrg4-6chr16:58463687-58463794(-)ndrg4-7chr16:58463740-58463826(+)
[0080]
表2.tap-qpcr中使用的引物及探针的命名及序列
[0081][0082][0083]
表3.tap-qpcr中使用的内参引物及探针的命名及序列
[0084]
名称荧光序列(5
‘‑3’
)actin_f/gagggaggaagtcatggcaggcttcactin_r/tcctggccaccttctcagccttaag
actin_pcy5agaaggcagcctgaagctggc
[0085]
在5%甲基化(使用经过亚硫酸盐转化后一代测序验证后、已知sdc2和ndrg4两基因启动子区域均 为完全甲基化的hct116细胞株,和后一代测序验证后已知sdc2和ndrg4两基因完全不甲基化的白细 胞dna进行混合,以sdc2和ndrg4两基因完全不甲基化的白细胞dna为稀释剂,稀释至hct116细 胞株dna量的比例为5%)的dna样本中,测试各单个候选甲基化检测引物探针组的检测能力。
[0086]
具体实验过程如下:
[0087]
1.dna提取
[0088]
根据dna提取试剂盒qiaamp dna blood mini kit说明书提取白细胞dna、hct116细胞株dna和 组织样品dna。
[0089]
2.tap转化
[0090]
使用enzymatic methyl-seq conversion module试剂盒中tet氧化体系及tet酶,按照试剂 盒说明书进行tet氧化反应,将甲基化胞嘧啶(5mc)、羟甲基化胞嘧啶(5
hm
c)氧化为5-羰基胞嘧啶(5-fc) 和5-羧基胞嘧啶(5-cac),然后通过吡啶硼烷反应将5-fc和5-cac还原为二氢尿嘧啶(dhu)。具体步 骤如下:
[0091]
1)tet氧化反应
[0092]
按照enzymatic methyl-seq conversion module试剂盒说明书进行,具体如下:
[0093]
a)将400μl tet2 reaction buffer加入到tet2 reaction buffer supplement中,混匀;
[0094]
b)将50ng前述5%甲基化dna加入ddh2o中配成29μl dna;
[0095]
c)在冰上,取16μl下表成分的混合物加入29μl dna中,混匀;
[0096][0097]
d)将1μl 500mm fe(ii)溶液加入到1249μl ddh2o中稀释,将稀释的fe(ii)溶液加入到步骤c的 dna中后,混匀;
[0098][0099]
e)在pcr仪中37℃孵育150分钟,加热盖子温度设置为45℃。
[0100]
f)将样品转移到冰上,加入2μl蛋白酶k终止。
[0101][0102]
g)上下吹打至少10次彻底混合,并短暂离心。
[0103]
h)在50℃处孵育60分钟,加热盖子设置为55℃,然后在4℃保存。
[0104]
2)tet氧化产物纯化
[0105]
a)提前30min将am pure xp beads磁珠拿出涡旋混匀平衡至室温。
[0106]
b)在位于pcr管中的步骤1)所得的52μl样品中加入104μl重新悬浮的am pure xp beads,上下至 少吹打10次,在最后一次混合过程中,小心地将所有的液体从尖端排出。
[0107]
c)在室温孵育5分钟。
[0108]
d)将pcr管放置在磁性支架上,静置5分钟(此时形成上清层和beads层),将beads从上清液中分离 出来,小心地弃去上清液,注意不要吸到beads。
[0109]
e)配制80%乙醇,在磁性支架上,将200μl 80%新制备的乙醇加入到pcr管中,在室温下孵育30秒, 然后小心地弃掉上清液,注意不要吸到beads。
[0110]
f)重复80%乙醇洗涤1次,弃去上清。
[0111]
g)打开ep管盖,干燥磁珠3分钟,注意不要过度干燥磁珠。
[0112]
h)从磁力架上取下pcr管,添加36μl ddh2o洗脱dna,上下吹打10次混匀,室温下孵育2分钟。
[0113]
i)把pcr管放在磁力架上,静置3分钟(或当溶液清澈时),将35μl上清液转移到新的pcr管中。
[0114]
3)吡啶硼烷还原反应
[0115]
a)通过调节4m的乙酸和5m的氢氧化钠溶液配置成3m的乙酸钠缓冲溶液(ph=4.3);
[0116]
b)将10μl的乙酸钠溶液、5μl 10m的吡啶硼烷加入到步骤2)所得的35μl dna样本中,37℃,850rpm 孵育16小时;
[0117]
4)吡啶硼烷反应后纯化
[0118]
根据zymo oligo clean&concentrator试剂盒说明进行纯化,具体如下:
[0119]
a)将完成步骤3)后的pcr管从加热混匀器中取出,将管壁液体离心至pcr管底。
[0120]
b)加入100μl oligo binding buffer。
[0121]
c)加400μl乙醇,涡旋混匀,短暂离心,将所有液体转移至spin
tm column,12000g离心30秒,弃掉 收集管中的废液。
[0122]
d)加750μl dna wash buffer至spin
tm column,12000g离心1分钟,弃掉收集管中的废液。
[0123]
e)将zymo-spin
tm column置于新的1.5ml去酶ep管中14000g离心1分钟,空离去除硅胶膜中残留乙 醇。
[0124]
f)将zymo-spin
tm column置于新的.5ml去酶ep管中,加25μl eb buffer洗脱dna,室温放置2分钟, 14000g离心1分钟。
[0125]
3.taps-qpcr
[0126]
使用kapa probe fast qpcr master mix(2x)kit按照如下反应体系加样,qpcr反应体系如下:
[0127]
[0128][0129]
qpcr反应条件如下:
[0130][0131]
4.检测结果
[0132]
1)见图4,显示了11对扩增子的扩增曲线,对应结果如下表4:
[0133]
表4
[0134][0135][0136]
图4和表4结果表明,引物设计和单重扩增方案设计成功。δct值越大,表明甲基化水平越高。对 于同一甲基化水平的样本,该δct值越大,表明该靶基因检测效率越高。
[0137]
本结果表明,各个扩增子设计成功。
[0138]
说明:白细胞dna甲基化水平为0;对于同一个5%甲基化dna样本,不同的检测标志物(即表4 所述靶基因),检测的δct差值不同。在内参基因ct值相同时,靶标基因ct值越小,则甲基化越强。因 此,靶标基因ct-内参基因ct这一差值越小,代表该靶基因的甲基化水平越强。
[0139]
实施例3:taps-qpcr单一标志物靶标检测肿瘤组织和正常组织样品
[0140]
将10例1期结直肠癌患者的肿瘤组织dna混在一起组成混合物,配对的正常组织dna混在一起组 成混合物,然后在肿瘤组织dna混合物和正常组织dna混合物中测试各单个候选甲基化检测引物探针组 的检测能力。
[0141]
实验操作等同于实施例2。
[0142]
如图5所示,sdc2-2、sdc2-1、sdc2-4、sdc2-3在normal mix中甲基化信号很弱(ct值在45左 右),在tumour mix中甲基化信号强(ct值在32左右),肿瘤组织的δct(内参基因ct均值1-靶标基 因ct均值1)和正常组织的δct(内参基因ct均值2-靶标基因ct均值2)相差较大,因此可以很好的区 分肿瘤组织和正常组织,sdc2-3在肿瘤组织中甲基化信号很强(ct值在29左右),但在正常组织中甲基 化信号也很强,背景信号太强。ndrg4-1、ndrg4-7、ndrg4-3、ndrg4-5、ndrg4-4、ndrg4-2肿瘤 组织的δct(内参基因ct均值1-靶标基因ct均值1)和正常组织的δct(内参基因ct均值1-靶标基因 ct均值1)相差较大,因此可以很好的区分肿瘤组织和正常组织,ndrg4-6在正常组织和肿瘤组织中均没 有检测到甲基化信号,认
为扩增失败。对应结果如下表5:
[0143]
表5
[0144][0145]
根据表5得知:δct值越大,甲基化水平越高。通过对肿瘤组织样品和正常组织样品的的检测,本发明 在后续应用中,删除了扩增效率最差的ndrg4-6靶基因。
[0146]
综上,图5给出了单个甲基化标志物肿瘤特异性测试结果,横轴为每一个标志物,纵轴越小表示该标 志物区分肿瘤组织和正常组织的能力越强。
[0147]
实施例4:tap-mqpcr多重检测方案用于不同甲基化成都样品的检测
[0148]
根据上述单一标志物测试的结果,选择能清晰区分肿瘤组织和正常组织的标志物sdc2-2、sdc2-1、 sdc2-4、ndrg4-1、ndrg4-7、ndrg4-3、ndrg4-5、ndrg4-4、ndrg4-2组成一个多重标志物测试 方法,多重测定中actb内参基因作为dna量的指标。
[0149]
实验方法
[0150]
1)、使用100%甲基化hct116细胞株和白细胞dna按照不同比例混合,制备甲基化程度不同的样 本(5%、2%、1%、0.5%、0.2%)进行tap-mqpcr检测限测试。
[0151]
以甲基化程度5%为例,就是将100%甲基化hct116细胞株与同浓度白细胞dna按照5:95的比例混 合。白细胞dna甲基化水平为0。
[0152]
2)、多重标志物测试方法中使用的标志物为:sdc2-2、sdc2-1、sdc2-4、ndrg4-1、ndrg4-7、 ndrg4-3、ndrg4-5、ndrg4-4、ndrg4-2,各标志物的引物及探针序列同实施例2的表2。
[0153]
3)、tap转化
[0154]
将模拟的不同甲基化比例的dna样本进行超声破碎及taps转化,具体实验步骤同实施例2的整个步 骤2(即步骤2的1)~4))。
[0155]
4)、tap-mqpcr
[0156]
4.1)、mqpcr引物和探针混合液准备:将单个靶标基因的引物和探针终浓度稀释为
5μm,内参基因 的引物和探针终浓度稀释为10μm。
[0157]
4.2)、qpcr反应体系如下:
[0158]
按照如下反应体系依次加入各成分,分装到每个反应孔中,在每个反应孔加入2μl经tap转化后的 dna样本。
[0159]
成分初始浓度终浓度体积/25μl反应(μl)kapa probe fast qpcr master mix2x1x12.59重引物混合物5μm0.3μm1.59重探针混合物5μm0.1μm0.5内参基因引物混合物10μm0.3μm0.75内参基因探针10μm0.1μm0.25去酶水 /7rox50x1x0.5dna模板(步骤3所得) 8ng/rxn2总体积 /25
[0160]
qpcr反应条件如下:
[0161][0162]
4.3)、实验结果
[0163]
见图6,扩增曲线的ct值随dna甲基化程度的降低而增大,不同甲基化dna(5%、1%、0.5%、0.2%、 buffy coat即白细胞)的扩增曲线从左到右依次排列,可以区分开。且0.2%甲基化样品具有明显的扩增信 号,且白细胞样品没有扩增出甲基化信号,表明本方法至少可以检测出甲基化程度低至0.2%的dna样品, 且其检测特异性较强,在白细胞样品中无假阳性信号。对应结果如下表6:
[0164]
表6
[0165][0166]
实施例5:单一甲基化标志物和多个甲基化标志物联合检测的灵敏度比较
[0167]
1.实验方法:
[0168]
1.1单一甲基化基因的的11套引物及探针见实施例2的表2。
[0169]
qpcr反应体系如下:
[0170][0171]
qpcr反应条件如下:
[0172][0173]
1.2多个甲基化标志物联合检测方法见实施例2描述的tap-mqpcr方法。
[0174]
2.实验结果
[0175]
见图7(单一甲基化标志物和多个甲基化标志物联合检测的灵敏度比较),多重标志物检测产生的δct 值高于单一标志物检测的δct,说明多个甲基化标志物联合检测较单一甲基化标志物检测信号更强。对应 结果如下表7:
[0176]
表7
[0177][0178]
实施例6:tap-mqpcr方法用于结直肠癌患者组织dna检测
[0179]
1.实验方法
[0180]
1)在温州医科大学附属第一医院入组10例i期结直肠癌肿瘤组织及配对的结直肠正常组织、10例进展 性腺瘤组织、10例良性息肉组织;
[0181]
2)使用dna提取试剂盒qiaamp dna mini kit,按照试剂盒中的说明书进行组织dna提取。
[0182]
3)提取的组织dna超声破碎为1kb左右,然后进行tap转化,具体实验步骤见实施例2的步骤2的 1.2、1.3。
[0183]
4)tap-mqpcr反应体系和反应条件见实施例2的步骤2的的1.4。
[0184]
2.实验结果
[0185]
入组40位受试者的临床特征及阳性检出率如下:
[0186][0187]
具体实验数据见表8;
[0188]
表8
[0189]
[0190][0191]
上表结果统计为图8,经过方差分析,正常组织样品与良性息肉、进展性腺瘤和肿瘤组织之间均有统 计学差异。
[0192]
结果经过统计和整理,如图8所示,纵轴δct越大表示甲基化水平越高,该方法能明显地区分正常组 织、肿瘤组织、良性息肉及进展性腺瘤组织,使用one-way anova统计学方法分析,良性息肉、进展性 腺瘤、肿瘤组织分别与与正常组织具有统计学差异。
[0193]
以δct为-10作为阈值,当满足δct》-10判断为阳性,δct≤-10判断为阴性。正常组织均为阴性,10 例良性息肉中检出8例阳性,10例进展性腺瘤均为阳性,10例肠癌组织中检出9例阳性。
[0194]
如图9所示,对正常组织和结直肠癌肿瘤组织进行受试者工作特征曲线(receiver operatingcharacteristic curve,roc曲线)分析,结果显示,该方法对crc肿瘤组织检测能力较强,曲线下面积 auc值为0.98,在特异性为100%情况下,敏感性为90%。
[0195]
结直肠癌的发生发展遵循正常组织、良性息肉、进展性腺瘤到肿瘤组织的过程,因
此,我们的检测结 果能够明显区分正常组织、良性息肉、进展性腺瘤和肠癌组织,因此可能作为结直肠异常组织的检测标志 物。
[0196]
实施例7:tap-mqpcr方案在结直肠癌患者粪便dna中的应用
[0197]
1.实验方法
[0198]
1)在温州医科大学附属第一医院入组19例结直肠癌粪便样本(ⅰ期5例,ⅱ期7例,ⅲ期4例,ⅳ期3 例)和14例健康人粪便样本
[0199]
2)按照《next-generation stool dna test accurately detects colorectal cancer and large adenomas》文献 中提及的粪便基因靶向捕获技术进行粪便人类基因组dna捕获。
[0200]
3)tap转化具体实验步骤同实施例2。
[0201]
4)tap-mqpcr检测甲基化水平,具体实验步骤同上述实施例。
[0202]
2.实验结果
[0203]
结直肠癌患者粪便样本tap-mqpcr检测结果见表9,如下:
[0204]
表9
[0205][0206]
健康人粪便dna样本tap-mqpcr检测结果见表10,如下:
[0207]
表10
[0208][0209]
根据该检测数据,如图10所示结直肠癌患者的粪便甲基化水平高于健康人,绘制受试者工作曲线 (receiver operating characteristic curve,roc曲线)。如图11所示,结果显示,曲线下面积值为0.605, 在特异性为85.71%情况下,敏感性为52.94%。
[0210]
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施 例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均 应认为是本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1