一种含三苯氧膦结构的咪唑型荧光分子及其制备方法和作为荧光探针的应用
1.本发明涉及一种荧光分子,具体涉及一种含三苯氧膦结构的咪唑型荧光分子,还涉及其制备方法以及作为检测fe
3+
的荧光探针应用,属于传感技术领域。
背景技术:
2.咪唑衍生物是指化合物结构中含有一个或多个咪唑环结构单元的一类物质的总称。咪唑是一个含有两个氮原子的五元环,属于1,3-唑类化合物,其化学性质稳定,广泛存在于天然产物与生物活性物质中。咪唑衍生物因其抗菌、消炎等药理特性在药物合成、蛋白酶活性抑、固化剂等方面均有报道,由于咪唑环反应位点多,所以被修饰改造出来的衍生物在光电材料、传感器和荧光探针等领域的研究也层出不穷。
3.近年来,研究者报道了大量以咪唑为基本单元所设计的材料,在室温磷光、深蓝色发光材料、力致变色、荧光传感和离子检测方面应用广泛。
[0004][0005]
如liu等成功开发出不同激发态的芘[4,5-d]咪唑衍生物,分别为pypa、pyppa、pyppac和pypac。其中,pyppa和pyppac具有赋予它们的混合局部和电荷转移状态、纯蓝色荧光以及高量子产率。基于pyppa的非掺杂oled cie坐标为(0.14,0.13),并达到最大eqe 8.47%。基于pyppac的非掺杂oled最大亮度为50046cd m
–2,位于蓝色区域,cie坐标为(0.15,0.21),eqe=6.74%,亮度超过10000cd m
–2。
[0006]
如wang等设计合成了一种新的吩嗪-咪唑基席夫碱(pis)荧光探针,已被开发用于在生理ph条件下对水性介质中的cd
2+
离子进行比率检测。
[0007]
[0008]
wang等共合成了三种新型双靶向荧光探针,并分别通过咪唑和一种半乳糖(im-gal-1)、两种半乳糖(im-gal-2)和三种半乳糖(im-gal-3)相连。这些探针对fe
3+
显示出良好的选择性和灵敏度,化学计量比为1:2识别模式。
[0009][0010]
目前,分子中同时含有咪唑和咔唑结构的有机发光分子也相继被设计合成出来。如文献(tagare j,dubey d k,yadav r a k,et al.triphenylamine-imidazole-based luminophores for deep-blue organic light-emitting diodes:experimental and theoretical investigations[j].materials advances,2020,1(4):666-679.);(devesing girase j,rani nayak s,tagare j,et al.solution-processed deep-blue(y~0.06)fluorophores based on triphenylamine-imidazole(donor-acceptor)for oleds:computational and experimental exploration[j].journal of information display,2021:1-15.);(bourouina a,rekis m.comparison in optoelectronic properties of triphenylamine-imidazole or imidazole as donor for dye-sensitized solar cell:theoretical approach[j].journal of molecular modeling,2021,27(8):1-9.)等报道了咪唑与三苯胺相连的深蓝色有机电致发光材料,但是,目前还未见同时含有咪唑和三苯氧膦结构的有机发光分子用于荧光探针检测铁离子的相关报道。
[0011]
现有技术中合成咪唑衍生物的方法也很多,如debus-radziszewski咪唑合成法是最经典的合成咪唑化合物的方法之一。二酮化合物和醛类化合物在胺或铵盐存在下,可以用来合成2,4,5-三芳基咪唑、菲并咪唑、双咪唑衍生物等。
[0012][0013]
sundar和rengan报道了以苯甲醇,1,2-二酮和过量乙酸铵为原料,进行三组分反应,在通入氧气的情况下,以0.25mol%的负载双钌(ii)催化该氧化过程,唯一的副产物是水。
[0014][0015]
wang等用各种双取代的炔烃、醛和胺为原料,通过新戊酸介导发生多组分反应,简便、高效地合成了四取代咪唑衍生物。
[0016][0017]
naidoo和jeena采用廉价的碘/二甲基亚砜(dmso)体系,通过“一锅、两步”法合成了2,4,5-三取代咪唑。该反应不需要借助酸性条件和金属,产率中等到良好。第一步是炔烃在i2/dmso条件下反应得到中间体:为二取代的苯偶酰,第二步醛和乙酸铵原位反应生成三取代的咪唑化合物。
[0018][0019]
grigg等通过异氰乙酸酯在银盐催化下均二聚化合成了咪唑化合物。在催化量的acoag条件下,异氰基乙酸甲酯与丙烯酸甲酯的反应形成吡咯啉,而在化学计量的acoag条件下,反应会发生异氰化物的均二聚化生成1,4-二取代咪唑。
[0020][0021]
shao等报道了基于酰胺基乙腈和苯硼酸的环化反应,形成了双取代的咪唑化合物。
[0022][0023]
xie等发明了一种新型的碱介导的活性亚甲基异氰酸酯与烯酮亚胺[3+2]环化反应。该反应具有区域选择性,烯酮亚胺在t-buok这种强碱的条件下与活性亚甲基异氰酸酯反应得到了1,4,5-三取代咪唑衍生物。
[0024][0025]
现有技术中合成咪唑的方法很多,并各自存在优点和缺点,目前为止还未见通过经典suzuki偶联和debus-radziszewski反应构建含三苯氧膦结构的咪唑环的小分子荧光探针的相关报道。
技术实现要素:
[0026]
针对现有技术存在的缺陷,本发明的第一个目的是在于提供一种含有三苯氧膦结构的咪唑型荧光分子,该分子物理和化学稳定性好,且在可见光和紫外光下表现出良好的荧光性能。
[0027]
本发明的第二个目的是在于提供一种有三苯氧膦结构的咪唑型荧光分子的制备方法,该方法原料易得,且合成步骤较为简单,产率可观,有利于扩大生产。
[0028]
本发明的第三个目的是在于提供一种有三苯氧膦结构的咪唑型荧光分子的应用,将其作为检测fe
3+
的荧光探针使用,荧光分子能够高选择性与fe
3+
结合并产生荧光淬灭,且受al
3+
、ca
2+
、li
+
、cu
2+
、na
+
、in
3+
等金属离子干扰小,可以用于fe
3+
的荧光检测,具有灵敏度高,检出限低等特点。
[0029]
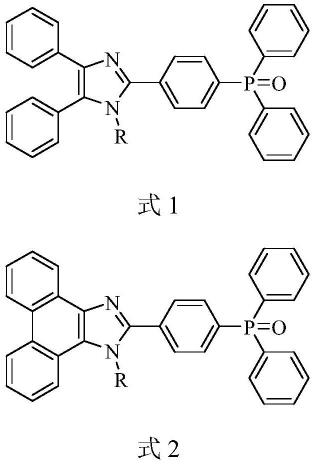
为了实现上述技术目的,本发明提供了一种含三苯氧膦结构的咪唑型荧光分子,其具有式1或式2结构:
[0030][0031]
其中,r为氢或芳基。
[0032]
作为一个优选的方案,所述芳基可以为苯基或者为苯基衍生的基团,如取代苯基,在苯环上还包含常见取代基团,具体如c1~c5的烷基、c1~c5的烷氧基、卤素取代基等等。本发明优选为最简单的苯环,可以延长咪唑环的共轭体系。
[0033]
本发明的荧光分子由咪唑结构与氧膦基通过苯环偶联形成典型的d-π-a结构,具有大共轭体系,刚性较强,其homo与lumo的能隙最大差距能(et)在3.71~3.85ev。从分子构型上看,氧膦基为电子给体(donor)、苯环为π桥、咪唑为电子受体(acceptor),氧膦基与多个苯基相连,增大了氧膦基的共轭性并改变了分子的空穴传输性能。此外,氧膦基的引入还能够提高荧光分子的溶解性能和热稳定性,如热稳定性均达到了250℃左右,有利于加工并拓宽其应用范围。
[0034]
本发明还提供了一种含三苯氧膦结构的咪唑型荧光分子的合成方法,该方法包括以下步骤:
[0035]
1)二苯基氧膦和对甲酰基苯硼酸通过suzuki偶联反应,得到4-二苯基膦酰基苯甲醛;
[0036]
2)4-二苯基膦酰基苯甲醛与苯偶酰或9,10-菲醌通过debus-radziszewski咪唑合成反应,即得式1或式2结构化合物。
[0037]
本发明通过两步经典的反应来合成含三苯氧膦结构的咪唑型荧光分子,先以二苯基氧膦与对甲酰基苯硼酸进行suzuki偶联反应,合成4-二苯基膦酰基苯甲醛,再利用4-二苯基膦酰基苯甲醛与二酮化合物和芳胺(或氨)进行经典的debus-radziszewski咪唑合成反应,得到兼具三苯氧膦结构和咪唑结构的荧光分子。
[0038]
作为一个优选的方案,二苯基氧膦和对甲酰基苯硼酸在1,4-二(二苯基膦基)丁烷、醋酸钯和碳酸钾作用下,于85~95℃温度下,反应20~24h。
[0039]
作为一个较优选的方案,二苯基氧膦和对甲酰基苯硼酸的摩尔比为1:1.5~1.6。
[0040]
作为一个较优选的方案,1,4-二(二苯基膦基)丁烷的摩尔量为二苯基氧膦摩尔量的4~6%。
[0041]
作为一个较优选的方案,醋酸钯的摩尔量为二苯基氧膦摩尔量的4~6%。
[0042]
作为一个较优选的方案,碳酸钾的摩尔量为二苯基氧膦摩尔量的0.4~0.6倍。
[0043]
作为一个优选的方案,4-二苯基膦酰基苯甲醛与苯偶酰或9,10-菲醌在乙酸铵和冰醋酸作用下,于115~125℃温度下,反应15~20h;或者,4-二苯基膦酰基苯甲醛与苯偶酰或9,10-菲醌及芳胺在乙酸铵和冰醋酸作用下,于115~125℃温度下,反应15~20h。
[0044]
作为一个较优选的方案,4-二苯基膦酰基苯甲醛与苯偶酰或9,10-菲醌等摩尔比。
[0045]
作为一个较优选的方案,乙酸铵与4-二苯基膦酰基苯甲醛的摩尔量比为2~3:1。
[0046]
作为一个较优选的方案,乙酸铵、芳基胺与4-二苯基膦酰基苯甲醛的摩尔量比为1~1.5:1~1.5:1。
[0047]
本发明的含三苯氧膦结构的咪唑型荧光分子的合成路线具体如下(四种目标荧光分子目标产物分别为c1、c2、c3和c4):
[0048][0049][0050]
本发明还提供了一种含三苯氧膦结构的咪唑型荧光分子的应用,其作为检测fe
3+
的荧光探针使用。本发明的含三苯氧膦结构的咪唑型荧光分子不但具有较好的荧光性能,
而且还能够与fe
3+
进行高选择性配位结合,fe
3+
能够使其荧光发生淬灭。利用该特点,可以实现fe
3+
的荧光检测,且具有检测灵敏度高,下限低等特点。
[0051]
作为一个优选的方案,含三苯氧膦结构的咪唑型荧光分子的应用于al
3+
、ca
2+
、li
+
、cu
2+
、na
+
、in
3+
中至少一种与fe
3+
共存体系中fe
3+
的荧光检测。本发明的含咔唑结构的咪唑型荧光分子对fe
3+
的作用选择性高,各类金属离子对铁离子的干扰小。
[0052]
相对现有技术,本发明技术方案带来的有益技术效果:
[0053]
本发明的荧光分子均具有较好的热稳定性,如荧光分子c1、c2、c3和c4,均达到了250℃左右。
[0054]
本发明的荧光分子具有较好的荧光性能,荧光分子c1和c2在可见光下呈纯白色,365nm紫外灯下显蓝色荧光,其中,c3和c4在可见光下呈浅黄色,365nm紫外灯下表现为蓝绿色荧光,c2的荧光量子产率最高(56.35%),c4的荧光寿命最长(5.1147ns)。
[0055]
本发明的荧光分子同时具有三苯氧膦结构和咪唑结构,荧光分子c1~c4均能与金属fe
3+
发生络合反应,使得分子的荧光受到抑制对铁离子表现出较高的选择性,其在溶剂中的荧光强度随着铁离子的加入而受到明显削弱,甚至淬灭,从而可以作为三价铁离子检测的荧光探针,其中c3表现出最佳的选择性,可用于定量检测fe
3+
。计算结果表明,4种分子的荧光强度与铁离子浓度均存在一定的线性关系,测得检出限lod分别为9.61
×
10
–5m、7.98
×
10
–5m、6.75
×
10
–5m和8.02
×
10
–5m。
[0056]
本发明的荧光分子合成方法和步骤简单,可以通过经典的suzuki偶联和debus-radziszewski咪唑合成反应等合成,且原料易得,产率可观,有利于扩大生产。
附图说明
[0057]
图1为荧光分子c1的1h nmr图谱。
[0058]
图2为荧光分子c1的
13
c nmr图谱。
[0059]
图3为荧光分子c1的h rms图谱。
[0060]
图4为荧光分子c1~c4的ftir光谱。
[0061]
图5为荧光分子c1~c4经归一化处理后的紫外可见吸收光谱。
[0062]
图6(a)为荧光分子c1~c4的固态荧光发射光谱;(b)为荧光分子c1~c4经归一化处理后的固态荧光发射光谱。
[0063]
图7(a)为荧光分子c1~c4的色度坐标;(b)为荧光分子c1~c4在自然光(i)和365nm紫外光(ii)下的照片。
[0064]
图8(a)为c1在五种不同极性溶剂(1
×
10
–5mol/l)中的pl光谱;(b)c2在五种不同极性溶剂中的pl光谱(1
×
10
–5mol/l)。
[0065]
图9(c)为c3在五种不同极性溶剂中的pl光谱(1
×
10
–5mol/l);(d)c4在五种不同极性溶剂中的pl光谱(1
×
10
–5mol/l)。
[0066]
图10为荧光分子c1~c4在不同离子溶液中的荧光发射光谱(1a,2a,3a,4a)和直方图(1b,2b,3b,4b)。
[0067]
图11为荧光分子c1~c4在365nm紫外线照射下的荧光图像;thf/h2o(fw=20%)溶液中的样品浓度为1
×
10
–5mol/l。
[0068]
图12(a)为c1在不同fe
3+
浓度溶液中的荧光光谱;(b)为c1的荧光发射强度随fe
3+
浓度的变化而变化;thf/h2o(fw=20%)溶液中的样品浓度为1
×
10
–5mol/l;fe
3+
的浓度为0~1
×
10
–2mol/l。
[0069]
图13为荧光分子c1~c4的cv曲线。
具体实施方式
[0070]
以下实施例旨在进一步详细说明本发明内容,而不是限制权利要求的保护范围。
[0071]
以下实施例中涉及的检测方法和仪器:
[0072]
用bruker-avance 400mhz核磁共振仪,测试1h(400mhz)和
13
c(100mhz)核磁共振谱,以cdcl3或dmso-d6为溶剂,以四甲基硅烷(tms)为内标,化学位移以ppm表示,所有耦合常数(j值)以赫兹(hz)表示。
[0073]
用perkin-elmer sp one红外分光光度计,进行红外光谱分析,采用溴化钾(kbr)压片。
[0074]
用wc-1型显微熔点仪测定所制备化合物的熔点,温度未经校正。
[0075]
用日立u-3100分光光度计测试紫外-可见吸收光谱(波长:200
–
800nm)。
[0076]
用hamamatsu稳态瞬态荧光光谱仪(edinburgh instruments,fls980),在325nm条件下的hecd激光激发下测试荧光光谱。
[0077]
在av1301a电化学工作站(上海辰华仪器有限公司)上进行循环伏安法(cv)测量,扫描速率为50mv/s,三个电极由铂丝工作电极、铂丝对电极和银/氯化银参比电极组成。
[0078]
以下实施例中涉及的化学药剂,如果没有特殊说明,均为常规的市售药品。
[0079]
实施例1
[0080]
(1)4-二苯基膦酰基苯甲醛的合成
[0081]
在氮气气氛下,将二苯基磷氧(5mmol)、4-甲酰基苯硼酸(7.5mmol)、1,4-二(二苯基膦基)丁烷(0.25mmol)、醋酸钯(0.25mmol)、碳酸钾(2.5mmol)和15ml二氧六环,依次加入到50ml带聚四氟乙烯支管口的烧瓶中,在90℃下反应24h。反应结束后,趁热用硅藻土过滤,再加饱和食盐水和二氯甲烷萃取,干燥得到粗产物,经柱层析分离纯化(洗脱剂为石油醚/二氯甲烷,体积比为10:1),得到白色固体,产率约为71%。1h nmr(400mhz,cdcl3):δ(ppm)=9.88(d,j=15.4hz,1h),7.78(s,2h),7.36
–
7.14(m,12h).
[0082]
(2)2-(4-二苯基膦酰基苯基)-4,5-二苯基-1h-咪唑(c1)的合成
[0083]
在氮气环境中,于150ml三颈烧瓶中加入苯偶酰(5mmol)、乙酸铵(10mmol)、4-二苯基膦酰基苯甲醛(5mmol)和冰醋酸(20ml),在120℃下反应18h。结束后静置冷却至室温,加入适量水,析出灰色固体。抽滤、水洗、干燥,得到粗产物。经柱层析分离纯化(洗脱剂为石油醚/乙酸乙酯,体积比为10:1),得到白色固体,产率为74%;熔点为239
–
240℃。1h nmr(400mhz,dmso-d6):δ(ppm)=12.94(s,1h),8.27
–
8.20(m,2h),7.79
–
7.62(m,8h),7.62
–
7.49(m,8h),7.46(t,j=7.3hz,2h),7.40(d,j=7.1hz,1h),7.31(t,j=7.4hz,2h),7.23(t,j=7.2hz,1h);
13
c nmr(100mhz,cdcl3):δ(ppm)=145.43,137.90,137.11,137.00,135.55,134.13,133.91,133.72,131.49,131.32,129.54,129.33,129.26,129.15,129.09,128.98,128.66,128.33,127.55,127.04,125.88,125.81;hrms calculated for c
33h26
n2op[m+h]
+
m/z 497.1777,found 497.1770.
[0084]
(3)2-(4-二苯基膦酰基苯基)-1,4,5-三苯基-1h-咪唑(c2)的合成
[0085]
在氮气环境中,于150ml三颈烧瓶中加入苯偶酰(5mmol)、乙酸铵(5mmol)、苯胺(5mmol)、4-二苯基膦酰基苯甲醛(5mmol)和冰醋酸(20ml),在120℃下反应20h。结束后静置冷却至室温,加入适量水,析出灰色固体。抽滤、水洗、干燥,得到粗产物。经柱层析分离纯化(洗脱剂为石油醚/乙酸乙酯,体积比为10:1),得到白色固体,产率约为78%;熔点为223
–
225℃。1h nmr(400mhz,cdcl3):δ(ppm)=7.60(ddd,j=31.2,17.8,8.1hz,1h),7.45(t,j=6.3hz,1h),7.33
–
7.27(m,1h),7.25
–
7.15(m,1h),7.12(d,j=6.9hz,1h),7.04(d,j=7.1hz,1h);
13
c nmr(100mhz,cdcl3):δ(ppm)=145.56,138.71,136.68,134.02,133.83,132.78,132.71,132.15,132.05,132.02,131.92,131.72,131.08,130.22,129.37,128.71,128.60,128.48,128.44,128.30,128.23,127.37,126.88;hrms calculated for c
39h30
n2op[m+h]
+
m/z 573.2090,found 573.2100.
[0086]
(4)2-(4-二苯基膦酰基苯基)-1h-菲并[9,10-d]咪唑(c3)的合成
[0087]
与c1合成方法类似,浅黄色固体,产率为58%;熔点为247
–
249℃。1h nmr(400mhz,cdcl3):δ(ppm)=8.70(t,j=8.4hz,1h),8.58(d,j=7.6hz,1h),8.43(d,j=6.6hz,1h),8.28(d,j=7.4hz,1h),7.88(dd,j=11.1,8.2hz,1h),7.83
–
7.40(m,7h);
13
c nmr(100mhz,cdcl3):δ(ppm)=160.84,145.24,135.62,132.73,132.63,132.26,132.20,132.10,131.59,130.63,129.55,128.96,128.77,128.65,127.53,127.36,126.99,126.87,126.77,126.37,126.00,123.76,123.44,122.92,120.95,120.82.hrms calculated for c
33h24
n2op[m+h]
+
m/z 495.1621,found 495.1602.
[0088]
(5)1-苯基-2-(4-二苯基磷酰基苯基)-1h-菲并[9,10-d]咪唑(c4)的合成
[0089]
与c2合成方法类似,淡白色固体,产率为55%;熔点为242
–
243℃。1h nmr(400mhz,dmso-d6):δ(ppm)=8.90(dd,j=20.8,8.3hz,2h),8.68(d,j=7.8hz,1h),7.88
–
7.48(m,19h),7.34(t,j=7.6hz,1h),7.07(d,j=8.2hz,1h),5.95(s,1h);
13
c nmr(100mhz,cdcl3):δ(ppm)=149.45,138.35,137.60,132.64,132.27,132.14,132.11,132.08,132.05,131.95,131.59,130.39,130.16,129.50,129.24,129.12,128.93,128.64,128.56,128.52,128.41,127.39,127.07,126.40,125.86,125.24,124.14,123.17,122.89,122.70,120.95;hrms calculated forc
39h28
n2op[m+h]
+
m/z 571.1934,found.571.1932.
[0090]
结构表征:
[0091]
对上述c1~c4四个咪唑分子进行了核磁共振氢谱、碳谱和傅里叶红外光谱的结构表征。4-二苯基膦酰基苯甲醛的氢谱,在化学位移值10.0ppm附近出现了醛基1号位氢的特征单峰,个数为1,且总的氢原子数与谱图中的总积分数相同,表明该醛被成功制备。c1的氢谱如图1所示,化学位移值13.0ppm附近出现了咪唑环中n
–
h键的氢单峰,8.25ppm附近为三苯氧膦结构中2号位苯环上氢的双峰;其余氢均在芳香区附近,氢原子总数与氢谱中剩余积分数相等。综合c1的碳谱图2、质谱图3:145.430ppm处的峰位为咪唑2号碳的特征峰,且总的碳峰数小于等于c1中相同化学环境的碳总数;质谱中分子加氢离子后理论值为497.1777,真实值为497.1770,符合质谱标准分子量误差范围,说明c1被成功合成。
[0092]
c1~c4的傅里叶红外光谱图,如图4所示。3440cm
–1处为c1和c3结构中咪唑n
–
h键的振动吸收峰,由于c1~c4中均含有苯环、氧膦双键和咪唑环碳氮双键等结构,于是它们有着相同的吸收峰,如:3051cm
–1附近为苯环的=c
–
h伸缩振动吸收峰,1603cm
–1为咪唑环中c=n特征吸收峰,1580cm
–1、1600cm
–1为苯环c=c吸收峰,1190cm
–1附近为p=o特征吸收峰。
[0093]
紫外光谱:
[0094]
将c1~c4分别溶于thf溶液中,配制成浓度均为1
×
10
–5mol/l的待测液,而后进行紫外可见吸收光谱测试,经过归一化处理后如图5所示;从图5中可以看到c1和c2有两个明显的吸收带,大致在240nm和330nm左右;化合物c3和c4有五个较为明显的吸收带,大致在250nm、260nm、335nm、345nm和365nm附近。240nm、250nm和260nm处的吸收带应为苯环基团的π
–
π*跃迁引起,后330nm附近吸收带属于咪唑的p
–
p*跃迁。c2较c1相比,330nm左右的吸收带蓝移了13nm左右,可能是咪唑n
–
h键被苯环取代后,分子的共轭程度更高的缘故;c4与c3相比较,330nm以上的三个波带吸收峰位置无明显变化,可能与它们的结构十分相似有关。
[0095]
固态荧光分析:
[0096]
通过稳态瞬态荧光光谱仪测试了c1~c4在固态下的荧光性质。图6(a)和图6(b)分别为固态下c1~c4荧光发射光谱图和其归一化图。图7(a)和图7(b)分别为其色坐标图和在自然光下或365nm紫外灯下固体照片,表1为固态下化合物c1~c4的激发波长、发射波长、荧光寿命和荧光量子产率等数据。由此可知,在可见光下除c4为淡黄色固体外,其余均为白色固体;在365nm紫外灯下c1~c4大致呈蓝绿色荧光,这与色坐标图中c1~c4坐标所在光区颜色一致,且发射波长均为400nm左右。其中,c1的固态荧光发射强度最大,为2.76
×
105(a.u.),c4的荧光寿命最长,为5.1147ns,c2的荧光量子产率最高为56.35%。由于c1和c2结构相近,c2在c1的基础上增加了一个苯环,同样地,c4比c3多引入了一个苯环,分别比较它们的荧光寿命和荧光量子产率可知,苯环的引入能提高相应化合物的荧光寿命和荧光量子产率(比如,c2的荧光量子产率为56.35%,c1的为17.62%;c4的荧光寿命为5.1147ns,c3的为0.4778ns)。这可能是苯环的引入,使得整个分子的电子在整个共轭体系中更加平均化,不仅增加了共轭程度,而且一定程度上调节了分子中的给受体能力。这为今后设计和调控性能更为优异的发光基团提供了很好的策略。
[0097]
表1.荧光分子c1~c4荧光数据
[0098][0099][0100]a固体粉末的最大激发波长.
[0101]b固体粉末的最大发射波长.
[0102]c固体粉末的荧光强度.
[0103]d固体粉末的荧光寿命.
[0104]e固体粉末的荧光量子产率.
[0105]
溶剂化效应:
[0106]
表2.c1~c4在五种溶剂中的最大发射波长a[0107][0108]a溶液浓度:1
×
10
–5mol/l。
[0109]
选取了溶解性较好的五种溶剂:dmf、丙酮、二氯甲烷、四氢呋喃和甲苯,分别对c1~c4进行了溶剂化效应分析。5种溶剂的最大吸收波长数据见表2,其测试液浓度均为1
×
10
–5mol/l,图8中(a)、(b)和图9中(c)、(d)分别对应着c1~c4在五种不同溶剂中的液态荧光发射的归一化光谱和液态荧光照片。分析可知:c1~c4均无明显的溶剂化变色效应,液态荧光均为蓝光,其中,c3的吸收波长随溶剂极性的增加有着逐渐增大的变化,即红移;没出现溶剂化效应的原因可能是c3在这五种溶剂中体现得不明显,但是就液态荧光来看,c3具备溶剂化效应的可能性不大。
[0110]
金属离子检测性能分析:
[0111]
(1)金属离子选择性检测:
[0112]
选用以氯离子作为阴离子、不同金属阳离子的无机盐,排除阴离子的干扰。含氯离子的7种无机盐:分别是alcl3、cucl2、licl、nacl、cacl2、incl3和fecl3,选用thf/h2o(fw=30%)的混合溶液为溶剂;以c1为例,先配制含有c1且样品浓度为的1
×
10-5
mol/l的四氢呋喃/水(fw=30%)的混合溶液3ml,后分别称量一定量的无机盐,分别加入到混合溶液中,使其金属离子浓度为1
×
10-2
mol/l,轻轻摇晃后置于超声波清洗仪中,设置超声功率为100%、时间定为30min,待金属离子与含样品的试液充分络合后,静置24h后马上进行液态荧光发射测试,确保含不同金属离子的测试液在同一电压、同一激发波长和同一批次下进行,c2~c3做法相同。
[0113]
c1~c4的荧光发射曲线如图10所示,结合它们在365nm紫外等下的液态荧光照片图11分析可知:它们的最大荧光发射强度在al
3+
、li
+
、na
+
、ca
2+
、in
3+
溶液中的荧光发射强度保持较高水平,甚至有的略带增强,在cu
2+
溶液中均存在一定的荧光抑制,不是特别明显,但在fe
3+
溶液中均存在很强的荧光抑制现象,荧光几近消失。这可能是因为含咪唑结构的c1和c3具有未被取代的n
–
h键,其n上的孤对电子可以和fe
3+
结合形成络合物,破坏了分子的共轭体系和分子间的聚集方式,从而削弱了c1和c3在四氢呋喃水溶液中的荧光。通过初步的离子筛选发现:c1和c3可以选择性感知fe
3+
。c2和c4具体原因不知,但均有作为fe
3+
荧光探针的潜力。
[0114]
(2)金属离子灵敏性检测:
[0115]
通过上一步金属离子选择性分析,发现fe
3+
对c1~c4的液态荧光强度具有很强的抑制作用,可以考虑将c1~c4应用于fe
3+
检测中。以c1为例,在一定的浓度范围内,配制了fe
3+
浓度分别为0、0.005
×
10-2
m、0.0125
×
10-2
m、0.02
×
10-2
m、0.0275
×
10-2
m、0.035
×
10-2
m、0.1
×
10-2
m和1
×
10-2
m的样品溶液,样品溶液为含c1的浓度为1
×
10-5
m的四氢呋喃/水溶液(fw=30%),待摇振均匀后,置于超声清洗仪中超声30min,功率定为100%,超声完毕后
再静置老化一天,随后立即进行不同浓度fe
3+
的液态荧光发射测试。
[0116]
根据stern
–
volmer方程:i0/i=1+k
sv
[fe
3+
],拟合它们的线性关系,其中i0和i分别是c1在最高荧光发射波长下不加fe
3+
和加fe
3+
后的荧光发射强度,[fe
3+
]代表铁离子浓度变化值,k
sv
是stern
–
volmer常数。然后根据公式lod=3σ/k计算,其中lod代表离子检出限,σ为c1不加fe
3+
时测量15次空白溶剂的荧光发射强度的标准偏差,k表示拟合的线性函数斜率,即为k
sv
值。
[0117]
如图12,c1对不同浓度的fe
3+
荧光猝灭响应存在规律性的变化,其荧光发射强度随离子浓度增加而呈现线性减弱。根据stern
–
volmer方程我们计算得出c1的lod为9.61
×
10-5
m,其线性方程为i0/i=1.6417+7770[fe
3+
](r2=0.9965),其中r2达到0.99左右,可以认为它具有了作为荧光探针的精度标准。其余c2~c4的线性方程拟合同上做法。
[0118]
电化学性能分析:
[0119]
以c1为例:以四丁基六氟磷酸铵为电解质、脱氧thf为溶剂,称取一定质量的c1,配成浓度为1mg/ml的待测液,待测液经过除氧处理后立即进行cv测试。选用ag/agcl作为参比电极,且氧化还原电位使用二茂铁
–
二茂铁校准(fc
–
fc
+
)氧化还原对作为外部标准;铂丝为辅助电极,玻碳电极为工作电极进行测试。先打开电脑主机、循环伏安测试软件,连接电化学工作站,待接通稳定后进行参数设定:扫描速率设定6.25mv/s,圈数定为4,电压范围经过测试不断优化。
[0120]
表3.荧光分子c1~c4的电化学性能
[0121][0122]atheonset of oxidation potentials relative to fc
–
fc
+
couple.
[0123]bestimated from absorption onset.
[0124]cdetermined from e
onset ox
.
[0125]destimated from e
onset ox and e
g.
[0126]
经过如此往复过程后得到c1~c4的循环伏安测试曲线图13。根据图和计算公式将其氧化及还原电位和电化学数据列入表3。用其氧化及还原电位和电化学数据列入表3。用eg=(1240/λ
起始
)(ev)、e
lumo
=(e
homo
+eg)(ev)公式计算最高分子轨道(homo)能级和最低分子轨道(lumo)能级,λ
起始
可由紫外可见吸收光谱的起始波长估计。
[0127]
通过上述计算公式得到:c1、c2、c3、c4的homo/lumo的结果分别为-4.74/-1.03ev;-5.32/-1.47ev;-5.25/-1.56ev;-5.29/-1.58ev。同样地,计算出了c1~c4的电化学能量间隙分别是3.71ev、3.85ev、3.69ev、3.71ev。相差不大,而且分别对比c1和c3,c2和c4可知,苯环的引入可以增大相似结构分子的能量间隙。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1