一种2-甲基-3-四氢呋喃硫醇的制备方法与流程
1.本发明涉及化学技术领域,尤其涉及一种2-甲基-3-四氢呋喃硫醇的制备方法。
背景技术:
2.现有工艺技术是用2,3-二氢呋喃为主要原料与溴素合成中间体2,3-二溴四氢呋喃,然后再与甲基格氏试剂继续反应合成产品,现有工艺中用到的2,3-二氢呋喃价格昂贵,不易采购,增加了生产制备的成本。
3.本发明将主要原料2,3-二氢呋喃更换为四氢呋喃,四氢呋喃较易采购,而且价格低,降低的生产成本,并且四氢呋喃更易于合成反应,从而提高了产品收率。2,3-二氢呋喃沸点54℃,而四氢呋喃沸点为66℃,沸点高了10℃,减少了溶剂的挥发,降低了原料损耗,节约了生产成本。并且四氢呋喃为主要原料合成出来的产品香气更好,得到了许多国内外大公司的认可,极大的开拓了国内外市场。
4.为此,我们提出一种2-甲基-3-四氢呋喃硫醇的制备方法。
技术实现要素:
5.本发明主要是解决上述现有技术所存在的技术问题,提供一种2-甲基-3-四氢呋喃硫醇的制备方法。
6.为了实现上述目的,本发明采用了如下技术方案,一种2-甲基-3-四氢呋喃硫醇的制备方法,包括以下工作步骤:
7.第一步:500ml三口圆底烧瓶内用氮气彻底置换,依次加入乙醚:60g,四氢呋喃:23g,降温至0℃,滴加溴素:50g,维持0~5℃滴加,滴毕,0~5℃保温搅拌半小时,做成中间体a:2,3-二溴四氢呋喃待滴加;
8.第二步:500ml三口圆底烧瓶内依次加入乙醚:150g;镁屑:12g,搅拌加热至27~30℃,向釜内通入少量溴甲烷启动反应后,控温30-35℃,维持回流状态下向釜内继续通溴甲烷,通毕搅拌半小时;
9.第三步:搅拌降温至-5℃,滴加步骤第一步制得的2,3-二溴四氢呋喃,控制滴加温度-5-0℃,滴加过程放热,历时2小时左右,滴加完毕,控温-5~0℃保温搅拌3小时后,滴入冰水进行水解,水解温度≤20℃,水解完毕,控温≤20℃,用盐酸中ph;
10.第四步:中和完毕,静止分层,水相用乙醚:150ml
×
3萃取三次,合并有机相、萃取相,水浴脱乙醚至内温70℃,然后油泵减压蒸出中间体b:2-甲基-3-溴四氢呋喃;
11.第五步:三口圆底烧瓶内依次加入n,n-二甲基甲酰胺:120g,碳酸钾:15.9g,搅拌降温至0℃下滴加硫代乙酸:18g,维持0~5℃滴加,历时2小时滴加完毕,然后0~5℃继续搅拌2小时;
12.然后取第四步骤制得的中间体b:2-甲基-3-溴四氢呋喃19g加入到上步骤的圆底烧瓶内,加热回流2小时,回流温度140~142℃,回流完毕,降至室温,抽滤,滤液蒸去n,n-二甲基甲酰胺后,加入纯水,用二氯甲烷萃取:50ml
×
4萃取四次,合并萃取相,脱溶后,再油泵
减压精制得到中间体c:2-甲基-3-乙酰硫基四氢呋喃;
13.第六步:500ml三口圆底烧瓶内依次加入纯:130g,氢氧化钾:12g,搅拌溶解后,维持5-10℃下滴加第五步骤中间体c:2-甲基-3-乙酰硫基四氢呋喃:16g,滴加完毕水浴升温至18~20℃搅拌保温3小时,保温完毕,降至20℃以下用盐酸中和ph值,然后分层,用二氯甲烷:50ml
×
4萃取四次,合并有机相与二氯甲烷相,脱二氯甲烷至内温70℃,用油泵减压精制得到产品2-甲基-3-四氢呋喃。
14.作为优选,所述第二步中向釜内通入少量溴甲烷启动反应时,溴甲烷的质量为5g。
15.作为优选,所述第二步中维持回流状态下向釜内继续通溴甲烷,溴甲烷的质量为48g。
16.作为优选,所述第三步中用盐酸中和至ph=4~5,盐酸的用量为24g。
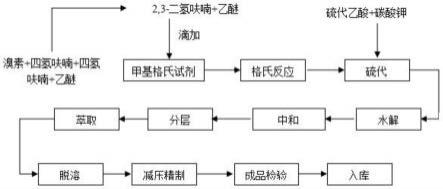
17.作为优选,所述第五步中滤液蒸去n,n-二甲基甲酰胺后,加入的纯水质量为100g。
18.作为优选,所述第六步中降至20℃以下用盐酸中和ph的值为2~3。
19.作为优选,所述第六步中用油泵减压精制得到产品2-甲基-3-四氢呋喃的收率达到85%以上。
20.作为优选,所述第三步中滴入冰水的用量为30g。
21.有益效果
22.本发明提供了一种2-甲基-3-四氢呋喃硫醇的制备方法。具备以下有益效果:
23.(1)、该一种2-甲基-3-四氢呋喃硫醇的制备方法,本发明解决了现有技术2,3-二氢呋喃价格昂贵,不易采购的问题,降低了生产制备的成本,本发明将主要原料2,3-二氢呋喃更换为四氢呋喃,四氢呋喃较易采购,而且价格低,降低的生产成本,并且四氢呋喃更易于合成反应,从而提高了产品收率,2,3-二氢呋喃沸点54℃,而四氢呋喃沸点为66℃,沸点高了10℃,减少了溶剂的挥发,降低了原料损耗,节约了生产成本,并且四氢呋喃为主要原料合成出来的产品香气更好,更容易获得市场的认可和青睐。
附图说明
24.为了更清楚地说明本发明的实施方式或现有技术中的技术方案,下面将对实施方式或现有技术描述中所需要使用的附图作简单的介绍。显而易见的,下面描述中的附图仅仅是示例性的,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图引伸获得其他的实施附图。
25.本说明书所绘示的结构、比例、大小等,均仅用以配合说明书所揭示的内容,以供熟悉此技术的人士了解与阅读,并非用以限定本发明可实施的限定条件,故不具技术上的实质意义,任何结构的修饰、比例关系的改变或大小的调整,在不影响本发明所能产生的功效及所能达成的目的下,均应仍落在本发明所揭示的技术内容得能涵盖的范围内。
26.图1为本发明2-甲基-3-四氢呋喃硫醇制备方法整体流程图;
具体实施方式
27.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他
实施例,都属于本发明保护的范围。
28.一种2-甲基-3-四氢呋喃硫醇的制备方法,如图1所示,第一步:500ml三口圆底烧瓶内用氮气彻底置换,依次加入乙醚:60g,四氢呋喃:23g,降温至0℃,滴加溴素:50g,维持0~5℃滴加,滴毕,0~5℃保温搅拌半小时,做成中间体a:2,3-二溴四氢呋喃待滴加;
29.第二步:500ml三口圆底烧瓶内依次加入乙醚:150g;镁屑:12g,搅拌加热至27~30℃,向釜内通入少量溴甲烷启动反应后,溴甲烷的质量为5g左右,控温30-35℃,维持回流状态下向釜内继续通溴甲烷,溴甲烷的质量约为48g,通毕搅拌半小时;
30.第三步:搅拌降温至-5℃,滴加步骤第一步制得的2,3-二溴四氢呋喃,控制滴加温度-5-0℃,滴加过程放热,历时2小时左右,滴加完毕,控温-5~0℃保温搅拌3小时后,滴入冰水进行水解,30g水解,水解温度≤20℃,水解完毕,控温≤20℃,用盐酸中和至ph=4~5,约用盐酸:24g;
31.第四步:中和完毕,静止分层,水相用乙醚:150ml
×
3萃取三次,合并有机相、萃取相,水浴脱乙醚至内温70℃,然后油泵减压蒸出中间体b:2-甲基-3-溴四氢呋喃;
32.第五步:三口圆底烧瓶内依次加入n,n-二甲基甲酰胺:120g,碳酸钾:15.9g,搅拌降温至0℃下滴加硫代乙酸:18g,维持0~5℃滴加,历时2小时滴加完毕,然后0~5℃继续搅拌2小时;
33.然后取第四步骤制得的中间体b:2-甲基-3-溴四氢呋喃19g加入到上步骤的圆底烧瓶内,加热回流2小时,回流温度140~142℃,回流完毕,降至室温,抽滤,滤液蒸去n,n-二甲基甲酰胺后,加入纯水,加入的纯水质量为100g,用二氯甲烷萃取:50ml
×
4萃取四次,合并萃取相,脱溶后,再油泵减压精制得到中间体c:2-甲基-3-乙酰硫基四氢呋喃;
34.第六步:500ml三口圆底烧瓶内依次加入纯:130g,氢氧化钾:12g,搅拌溶解后,维持5-10℃下滴加第五步骤中间体c:2-甲基-3-乙酰硫基四氢呋喃:16g,滴加完毕水浴升温至18~20℃搅拌保温3小时,保温完毕,降至20℃以下用盐酸中和ph值,ph的值为2~3,然后分层,用二氯甲烷:50ml
×
4萃取四次,合并有机相与二氯甲烷相,脱二氯甲烷至内温70℃,用油泵减压精制得到产品2-甲基-3-四氢呋喃,收率达到85%以上。
35.本发明的反应原理:
[0036][0037]
本发明解决了现有技术2,3-二氢呋喃价格昂贵,不易采购的问题,降低了生产制备的成本,本发明将主要原料2,3-二氢呋喃更换为四氢呋喃,四氢呋喃较易采购,而且价格低,降低的生产成本,并且四氢呋喃更易于合成反应,从而提高了产品收率,2,3-二氢呋喃沸点54℃,而四氢呋喃沸点为66℃,沸点高了10℃,减少了溶剂的挥发,降低了原料损耗,节约了生产成本,并且四氢呋喃为主要原料合成出来的产品香气更好,更容易获得市场的认可和青睐。
[0038]
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1