基于二代测序技术的高通量大型真菌分子鉴定方法与流程
1.本发明属于二代高通量测序及数据分析领域,具体涉及一种基于二代测序技术的高通量大型真菌分子鉴定方法。
背景技术:
2.在大型真菌野外资源调查鉴定及系统发育研究中,传统的方法一般是通过提取单个大型真菌子实体样品的基因组dna,并对其核糖体基因转录间隔区(its)序列进行pcr扩增,随后利用sanger测序技术对获取的pcr产物进行测序,最后人工逐一对双向序列进行拼接以获取its序列信息,然而对于大部分大型真菌子实体样品而言,由于其自身具有多个拷贝或与其他真菌存在寄生、伴生或共生等关系,导致获取的its基因片段不纯,进而导致测序过程容易出现双峰而造成测序失败。
3.对于此类样品需要将其its基因片段通过ta克隆技术连接至t载体,并经转化、单克隆菌落挑取、摇瓶培养、质粒提取等步骤后才能利用sanger测序技术对其its序列进行测序。对于大批量的大型真菌样本来说,其测序成本较高,且测序失败的样品,还需要进一步通过ta克隆来完成测序,实验繁杂,工作量庞大,工作周期长。
4.二代测序又称为高通量测序,具有测序成本低、深度深、数据量大等优势,在样品检测方面有巨大的潜力和应用价值。扩增子(its或16s)测序是高通量测序应用的主要技术之一,是一种对特定pcr产物或捕获片段进行测序的高靶向性方法,可用于分析特定环境中的目标区域。
5.当前,扩增子测序被广泛应用于细菌、真菌、古菌等物种分类以及土壤、海洋、肠道微生物等研究中,具有对模板要求低、操作简单、特异性强、灵敏度高、测序时间短等优点;相较于454,iontorrent等高通量测序平台而言,illuminahiseq测序平台是一类测序通量更高、成本更低的短读长测序平台,其测序长度可以满足对于大型真菌分类鉴定工作的开展。
6.与传统的sanger测序法相比,基于扩增子富集的二代高通量测序技术具有测序成本低、结果准确率高和实验强度小等优点,因此该技术的应用越来越广泛,但目前仍缺少利用二代高通量测序技术批量化鉴定大型真菌样本的方法或工具。
技术实现要素:
7.有鉴于此,本发明的主要目的在于提供一种基于二代测序技术的高通量大型真菌分子鉴定方法。
8.为达到上述目的,本发明的技术方案是这样实现的:
9.本发明实施例提供一种基于二代测序技术的高通量大型真菌分子鉴定方法,该方法为:
10.所以提取的大型真菌基因组dna为模板,通过pcr扩增its1及its2序列,获得pcr产物;
11.以所述pcr产物为模板,通过特异引物序列组合为its1及its2序列添加barcode标签序列,获得带有barcode标签序列的its1及its2pcr产物;
12.将所有带有barcode标签序列的its1及its2pcr产物分别进行混合,之后经切胶回收、建库后,获得混合pcr产物样品;
13.通过测序平台illumina hiseq pe250对所述混合pcr产物样品进行双端测序,获得高通量测序结果;
14.对所述高通量测序结果进行adapter序列的预测及去除、序列拆分、barcode标签序列的删除、序列拼接及物种注释。
15.上述方案中,所述通过pcr扩增its1及its2序列中,pcr的反应程序为:95℃3min;95℃20s,47℃20s,72℃30s,35个循环;72℃5min;4℃保存。
16.上述方案中,所述通过特异引物序列组合为its1及its2序列添加barcode标签序列中,pcr的反应程序为:95℃3min;95℃20s,47℃20s,72℃30s,10个循环;95℃20s,59℃20s,72℃30s,25个循环;72℃5min;4℃保存。
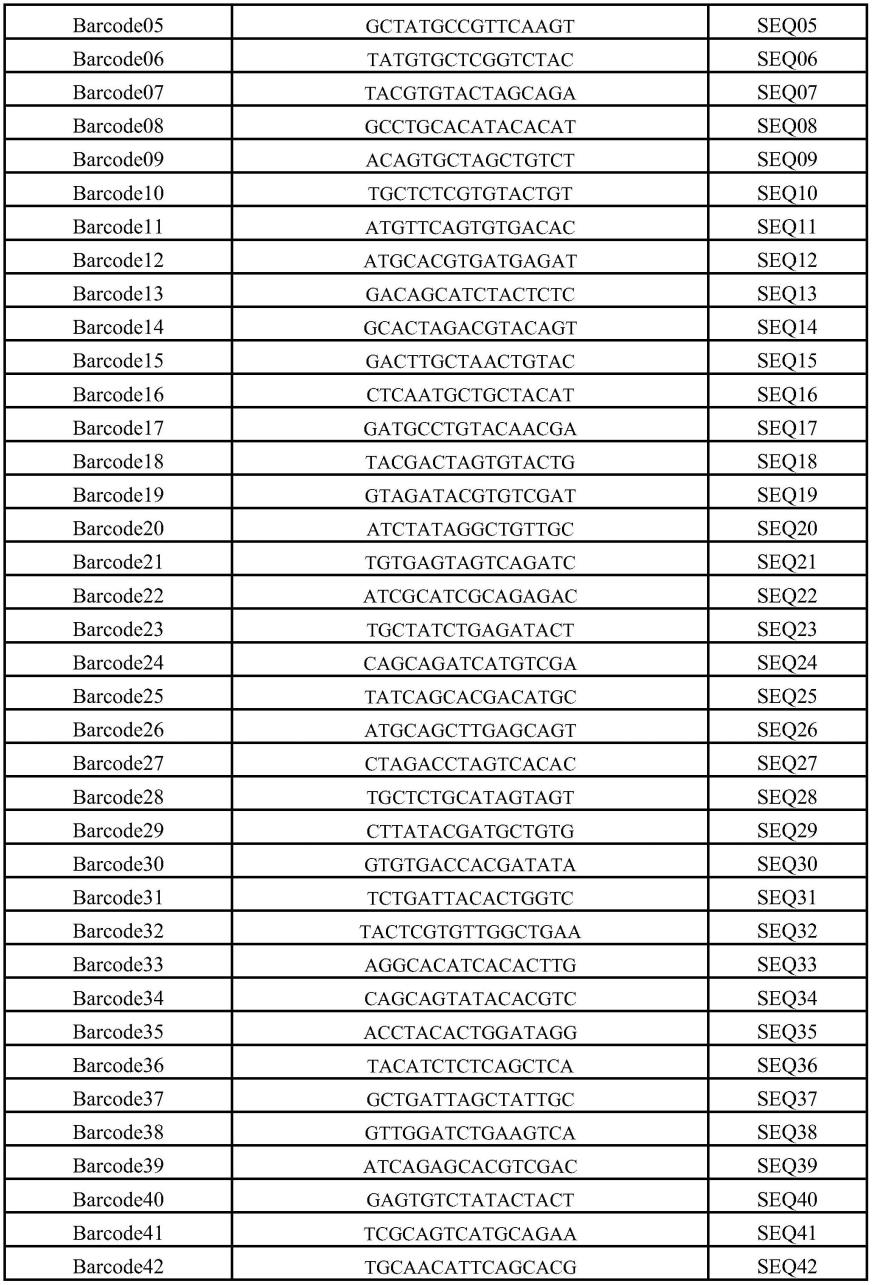
17.上述方案中,所述barcode标签序列如表所示:
18.19.[0020][0021]
上述方案中,所述通用引物序列为acchgcggarggatcattac、ttydcnrcgttcttcatcgt、gtgavtcatcrartntttga或者cctbcscttantdatatgcc。
[0022]
上述方案中,所述带有barcode标签序列的its1产物的序列如表所示:
[0023][0024]
上述方案中,所述带有barcode标签序列的its2产物的序列如表所示:
[0025][0026]
上述方案中,所述对所述高通量测序结果进行adapter序列的预测及去除、序列拆分、barcode标签序列的删除、序列拼接及物种注释,具体为:
[0027]
adapter序列的预测及去除:使用fastp预测并去除adapter序列,滑动窗口的长度设置为16;
[0028]
序列拆分:按照每个大型真菌样品特有的barcode标签序列,使用fastq-multx对混合样品的测序结果进行序列拆分,将拆分后的序列文件按样品id放到不同的文件夹中,并分别构建manifest;
[0029]
barcode标签序列的删除及序列拼接:使用dada2分别对每种大型真菌样品序列进行barcode标签序列的删除及序列拼接;
[0030]
物种注释:使用训练后的注释器对所有序列进行物种注释。
[0031]
上述方案中,所述注释器是以添加大量大型真菌its序列信息的unite数据库作为训练集,使用贝叶斯分类算法,经训练后所获得。
[0032]
与现有技术相比,本发明一次性可完成最多2500(50
×
50)个大型真菌样品的分子鉴定,相较于对每个样品单独进行一代sanger测序,大大降低了实验成本;成功率高:避免
了一代测序因its序列片段不纯而引导致的测序失败(双峰);避免了因测序失败(双峰)而进行的ta克隆带来的繁杂的工作,减少了实验步骤,节约实验材料,缩短了实验周期。
具体实施方式
[0033]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0034]
需要说明的是,在本文中,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、物品或者装置不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、物品或者装置所固有的要素。在没有更多限制的情况下,由语句“包括一个
……”
限定的要素,并不排除在包括该要素的过程、物品或者装置中还存在另外的相同要素。
[0035]
本发明实施例提供一种基于二代测序技术的高通量大型真菌分子鉴定方法,该方法通过以下步骤实现:
[0036]
步骤1:以提取的大型真菌基因组dna为模板,通过pcr扩增its1及its2序列,获得pcr产物;
[0037]
具体地,所述通过pcr扩增its1及its2序列中,pcr的反应程序为:95℃3min;95℃20s,47℃20s,72℃30s,35个循环;72℃5min;4℃保存。
[0038]
以its1(its1f:5
’‑
acchgcggarggatcattac-3’;its2r:5
’‑
ttydcnrcgttcttcatcgt-3’)和its2(gits7f:5
’‑
gtgavtcatcrartntttga-3’;its4r:5
’‑
cctbcscttantdatatgcc-3’)通用引物作为扩增引物,使用购买自vazyme(南京诺唯赞生物科技有限公司)的rapid taq master mix酶进行pcr扩增。
[0039]
pcr扩增体系为20μl,由10μl2
×
rapid taq master mix、1μl上游引物、1μl下游引物、1μl dna、7μl ddh2o组成。该反应体系中,dna为不同大型真菌样本,引物的浓度为10μm。
[0040]
步骤2:以所述pcr产物为模板,通过特异引物序列组合为its1及its2序列添加barcode标签序列,获得带有barcode标签序列的its1及its2 pcr产物;
[0041]
具体地,pcr的反应程序为:95℃3min;95℃20s,47℃20s,72℃30s,10个循环;95℃20s,59℃20s,72℃30s,25个循环;72℃5min;4℃保存。
[0042]
pcr扩增体系为30μl,由15μl 2
×
rapid taq master mix、1.5μl上游引物、1.5μl下游引物、2μl第一轮pcr产物、10μlddh2o组成。
[0043]
所述barcode标签序列如表所示:
[0044]
[0045]
[0046]
[0047][0048]
所述通用引物序列为acchgcggarggatcattac、ttydcnrcgttcttcatcgt、gtgavtcatcrartntttga或者cctbcscttantdatatgcc。。
[0049]
所述带有barcode标签序列的its1产物的序列如表所示:
[0050][0051]
所述带有barcode标签序列的its2产物的序列如表所示:
[0052]
步骤3:将所有带有barcode标签序列的its1及its2pcr产物分别进行混合,之后经切胶回收、建库后,获得混合pcr产物样品;
[0053]
步骤4:通过测序平台illumina hiseq pe250对所述混合pcr产物样品进行双端测序,获得高通量测序结果;
[0054]
步骤5:对所述高通量测序结果进行adapter序列的预测及去除、序列拆分、barcode标签序列的删除、序列拼接及物种注释。
[0055]
具体地,adapter序列的预测及去除:使用fastp预测并去除adapter序列,滑动窗口的长度设置为16;
[0056]
序列拆分:按照每个大型真菌样品特有的barcode标签序列,使用fastq-multx对混合样品的测序结果进行序列拆分,将拆分后的序列文件按样品id放到不同的文件夹中,并分别构建manifest;
[0057]
barcode标签序列的删除及序列拼接:使用dada2分别对每种大型真菌样品序列进行barcode标签序列的删除及序列拼接;
[0058]
物种注释:使用训练后的注释器对所有序列进行物种注释。
[0059]
所述注释器是以添加大量大型真菌its序列信息的unite数据库作为训练集,使用
贝叶斯分类算法,经训练后所获得。
[0060]
实施例1:
[0061]
在本实施例中,以100份大型真菌样本为例(样本编号分别为m001-m100),利用本发明提供的barcode特异性引物标记进行二代高通量测序,具体过程如下:
[0062]
(1)大型真菌样本dna提取
[0063]
dna的提取选用ezup柱式真菌基因组dna抽提试剂盒(上海生工生物工程股份有限公司),操作方法参照试剂盒提取说明书进行。
[0064]
(2)第一轮pcr反应
[0065]
通过第一轮pcr反应实现大型真菌样本its1和its2目标区域的富集。以上述提取的大型真菌样本dna为模板,分别以its1(its1f:5
’‑
acchgcggarggatcattac-3’;its2r:5
’‑
ttydcnrcgttcttcatcgt-3’)和its2(gits7f:5
’‑
gtgavtcatcrartntttga-3’;its4r:5
’‑
cctbcscttantdatatgcc-3’)通用引物作为扩增引物,使用购买自vazyme(南京诺唯赞生物科技有限公司)的rapid taq master mix酶进行pcr扩增。
[0066]
pcr扩增体系为20μl,由10μl 2
×
rapid taq master mix、1μl上游引物、1μl下游引物、1μl dna、7μl ddh2o组成。该反应体系中,dna为不同大型真菌样本,引物的浓度为10μm。
[0067]
反应程序:95℃3min;95℃20s,47℃20s,72℃30s,35个循环;72℃5min;4℃保存。
[0068]
进行1%琼脂糖凝胶电泳,调节电压120v,电泳25min,利用凝胶成像系统观察电泳结果。
[0069]
(3)barcode标签序列的添加
[0070]
以第一轮pcr反应产物作为模板,使用特异引物序列组合进行pcr扩增,进行第二轮pcr扩增,为its序列添加barcode标签序列,如下表所示。
[0071]
[0072][0073]
pcr扩增体系为30μl,由15μl 2
×
rapid taq master mix、1.5μl上游引物、1.5μl下游引物、2μl第一轮pcr产物、10μl ddh2o组成。
[0074]
反应程序:95℃3min;95℃20s,47℃20s,72℃30s,10个循环;95℃20s,59℃20s,72℃30s,25个循环;72℃5min;4℃保存。
[0075]
(4)混样及pcr产物纯化
[0076]
将所有样品的第二轮pcr产物混合,进行1%琼脂糖凝胶电泳,调节电压为120v,电泳25min,利用凝胶成像系统观察电泳结果,切下含有目的片段的凝胶,用上海生工生物工程股份有限公司的sanprep柱式dna胶回收试剂盒回收目的片段,具体操作步骤参照说明书。使用onedrop-1000检测回收产物的浓度与纯度,于-20℃冰箱中保存备用。
[0077]
(5)建库测序
[0078]
将上述pcr回收产物送至北京擎科生物科技有限公司西安分公司进行建库测序,使用illumina hiseq pe250测序平台分别对混合pcr产物样品进行双端测序,样品的测序深度为3g。
[0079]
(6)使用编写的脚本自动对高通量测序结果进行处理,具体如下:
[0080]
①
使用fastp预测并去除adapter序列,滑动窗口的长度(-w)设置为16;
[0081]
②
序列拆分:按照每个大型真菌样品特有的barcode标签序列,使用fastq-multx对混合样品的测序结果进行序列拆分,将拆分后的序列文件按样品id放到不同的文件夹中,并分别构建manifest。
[0082]
③
barcode标签序列的删除及序列拼接:使用dada2分别对每种大型真菌样品序列进行barcode标签序列的删除及序列拼接。
[0083]
④
物种注释:使用训练后的注释器对所有序列进行物种注释,其中,注释器是以添加大量大型真菌its序列信息的unite数据库作为训练集,使用贝叶斯分类算法,经训练后所获得。
[0084]
(7)数据整理
[0085]
整理分子鉴定结果。
[0086]
对比例1:
[0087]
使用一代测序技术对样品m001-m100进行测序,具体步骤如下:
[0088]
以提取的大型真菌样本dna作为模板,以its通用引物(its4:5
’‑
tcctccgcttattgatatg-3’;its5:5
’‑
ggaagtaaaagtcgtaacaagg-3’)作为扩增引物,使用购买自vazyme(南京诺唯赞生物科技有限公司)的rapid taq master mix酶进行pcr扩增。
[0089]
pcr扩增体系为20μl,由10μl 2
×
rapid taq master mix、1μl上游引物、1μl下游引物、1μl dna、7μl ddh2o组成。该反应体系中,dna为不同大型真菌样本,引物的浓度为10μm。
[0090]
反应程序:95℃3min;95℃20s,48℃20s,72℃30s,35个循环;72℃5min;4℃保存。
[0091]
将pcr产物送至北京擎科生物科技有限公司西安分公司进行一代sanger测序,测序引物为pcr扩增时所使用的引物,测序要求为双向测通,将测序成功的序列逐一进行拼接获得大型真菌样本its序列。
[0092]
将上述一代sanger测序所获的its序列上传至ncbi blastn进行序列比对,获取大型真菌分子鉴定结果(具体见下表)。
[0093][0094]
以上所述,仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1