一种肟脂化合物及其制备方法及应用
1.本发明属于有机合成及应用领域,具体涉及一种肟脂化合物及其制备方法及应用。
背景技术:
2.在有机合成化学中,利用光催化产生的活性物质具有优秀的催化能力和应用广泛性,这些活性物质如自由基、碱、酸等,在催化烯烃、炔烃加成反应和巯烯/炔点击化学反应等方面得到广泛的应用。由于光催化反应的高效性,避免使用大量的热催化剂和金属试剂,降低了对反应体系的要求和环境污染,特别是在多官能团的敏感化合物的制备中,开发新型的光催化剂具有极高的应用价值。
3.现有研究报道过的肟脂类化合物存在可见光吸收不够好、性质不太稳定、成本较高等问题。
技术实现要素:
4.本发明的目的在于提供一种具有可见光吸收好、性质稳定、成本低等优势的肟脂化合物。
5.为实现上述目的,本发明采用的技术方案如下:一种肟脂化合物,其结构式为:其中,r1为-h或-ch3;r2为-ch3或-ph。
6.本发明还提供上述肟脂化合物的制备方法,该方法简单、经济。
7.所述肟脂化合物的制备方法,包括以下步骤:(1)将含有羧酸化合物的二氯甲烷或四氢呋喃溶液与乙二酰二氯或二氯亚砜先在冰水浴中密封搅拌反应1小时,再缓慢升至室温;所述的羧酸化合物为2-乙酸基-9h-硫杂蒽-9-酮或2-丙酸基-9h-硫杂蒽-9-酮中的任意一个,其中:羧酸化合物与二氯甲烷或四氢呋喃用量比为1mmol∶1ml,羧酸化合物和乙二酰二氯或二氯亚砜的加料摩尔比为1∶1.5—20;然后由薄层色谱法观察反应;(2)上述反应结束后,母液在避水、避光条件下蒸干溶剂,得到白色产物;(3)向上述白色产物中加入含有肟的二氯甲烷溶液和三乙胺在冰水浴中搅拌反应6—8小时,其中,肟和步骤(1)中羧酸化合物的投料摩尔比为1∶1;所述的肟为苯乙酮肟或丙酮肟中的任意一个;(4)步骤(3)得到的反应溶液经9倍体积比的石油醚沉淀得到肟脂化合物粗产品,后经体积比为5∶1的石油醚/乙酸乙脂混合溶剂重结晶提纯得到肟脂化合物。
8.步骤(1)中所述的薄层色谱法观察反应的条件是:毛细管取样反应母液并用1滴无水甲醇稀释后再点样,以紫外光照法显色,其中羧酸原料点比移值小于0.3,产物原料点比
移值大于0.9,反应结束,展开体系为二氯甲烷/甲醇,其体积比为3∶1。
9.步骤(3)中的二氯甲烷和肟的用量关系为20—40ml/g。
10.步骤(4)中的石油醚/乙酸乙脂混合溶剂和肟脂化合物粗产品的用量关系为7—10ml/g。
11.步骤(3)得到的反应溶液浓缩后经石油醚/乙酸乙脂混合溶剂为展开剂进行柱层析分离得到所述肟脂化合物,其中,石油醚/乙酸乙脂混合洗脱剂的体积比为3∶1。
12.本发明提供了所述肟脂化合物在可见光催化条件下产生自由基应用于催化烯烃加成反应。
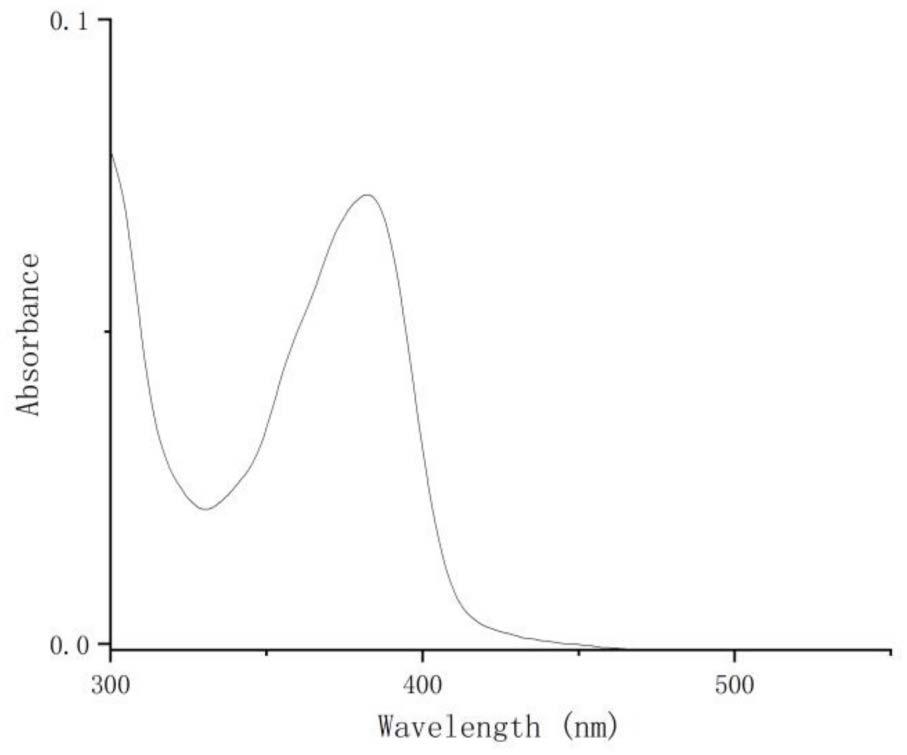
13.本发明的有益技术效果在于:(1)本发明中的肟脂化合物具有较大的共轭结构,较宽的光谱吸收范围(200到500纳米),在可见光范围内有较强吸收;(2)本发明中的肟脂化合物制备方法简单,产品价格低、分子小,具有良好溶解能力,与其它蒽酮类化合物相比具有较好的溶解性,适用范围更广;(3)本发明中的肟脂化合物光照条件下同时产生蒽酮苄基自由基,比一般的芳自由基和脂肪族自由基催化活性高。
附图说明
14.图1为本发明实施例1所得肟脂化合物的紫外-可见光谱图;图2为本发明实施例1所得肟脂化合物的核磁共振氢谱图;图3为本发明实施例1所得肟脂化合物的核磁共振碳谱图;图4为本发明实施例2所得肟脂化合物的核磁共振氢谱图;图5为本发明实施例2所得肟脂化合物的核磁共振碳谱图;图6为本发明实施例3所得肟脂化合物的核磁共振氢谱图;图7为本发明实施例3所得肟脂化合物的核磁共振碳谱图;图8为含本发明实施例1的肟脂化合物催化烯烃聚合应用的实时红外双键转化率曲线图;图9为本发明实施例1所得的肟脂化合物的苯溶液在405纳米光源照射下产生自由基的电子自旋共振谱图。
具体实施方式
15.下面结合附图和实施例,对本发明进行具体描述。
16.实施例1(1)将含有2-乙酸-9h-硫杂蒽-9-酮的四氢呋喃溶液与二氯亚砜在冰水浴中密封搅拌反应1小时,再缓慢升至室温反应(不高于25度)。其中:2-乙酸-9h-硫杂蒽-9-酮与四氢呋喃用量比为1mmol∶1ml,2-乙酸-9h-硫杂蒽-9-酮和二氯亚砜的加料摩尔比为1∶1.5—5;经薄层色谱法跟踪反应进行,毛细管取样反应母液并用1滴无水甲醇稀释后再点样,以紫外光照法显色,其中羧酸原料点比移值小于0.3,产物原料点比移值大于0.9,反应结束,展开体系为二氯甲烷/甲醇,其体积比为3∶1,取样的反应母液须用甲醇猝灭;(2)上述反应结束后,母液在避水、避光条件下蒸干溶剂(避免使用循环水真空泵
j = 8.2, 1.5 hz, 2h), 7.76
ꢀ–ꢀ
7.46 (m, 5h), 4.40 (q, j = 7.0 hz, 1h), 1.98 (s, 3h), 1.78 (s, 3h), 1.70 (d, j = 7.0 hz, 3h)。
23.所得肟脂的核磁共振碳谱图见图5;其中,
13
c nmr (101 mhz, cdcl3) δ 179.74, 174.43, 138.86, 137.14, 136.09, 132.25, 131.63, 129.83, 129.16, 129.08, 128.75, 126.42, 126.29, 126.08, 125.98, 52.22, 45.11, 18.39。
24.所得肟脂的高分辨质谱数据为:ultrflextof/tof (m/z)∶ c
18h15
no3s
+
,理论值325.0773,实测值325.0765 [m]
+
。
[0025]
实施例3(1)将含有2-丙酸-9h-硫杂蒽-9-酮的四氢呋喃溶液与乙二酰二氯在冰水浴中密封搅拌反应1小时,再缓慢升至室温反应(不高于25度)。其中:2-丙酸-9h-硫杂蒽-9-酮与乙二酰二氯用量比为1mmol∶1ml,2-丙酸-9h-硫杂蒽-9-酮和乙二酰二氯的加料摩尔比为1∶ 1.5—10;经薄层色谱法跟踪反应进行,其中羧酸原料点比移值小于0.3,产物原料点比移值大于0.9,反应结束,展开体系为二氯甲烷/甲醇,其体积比为3∶1,取样的反应母液须用甲醇猝灭;(2)上述反应结束后,母液在避水、避光条件下蒸干溶剂,得到白色产物不经提纯直接进行下一步反应;(3)向上述白色产物中加入含有苯乙酮肟的二氯甲烷溶液和三乙胺在冰水浴中搅拌过夜(7小时),其中,苯乙酮肟和2-丙酸-9h-硫杂蒽-9-酮的投料摩尔比为1∶1,二氯甲烷和肟的用量关系是30ml/g;(4)反应溶液经9倍体积比的石油醚沉淀得到肟脂化合物粗产品,后经体积比为5∶1的石油醚/乙酸乙脂混合溶剂重结晶提纯得到肟脂化合物(产率93%),混合溶剂和粗品的用量关系是9ml/g。
[0026]
所得肟脂的结构式为:所得肟脂的核磁共振氢谱图见图6;其中,1h nmr (400 mhz, cdcl3) δ 8.67
ꢀ–ꢀ
8.58 (m, 2h), 7.74
ꢀ–ꢀ
7.37 (m, 10h), 4.13 (q, j = 7.1 hz, 1h), 2.28 (s, 3h), 1.71 (d, j = 7.2 hz, 3h)。
[0027]
所得肟脂的核磁共振碳谱图见图7;其中,
13
c nmr (101 mhz, cdcl3) δ 179.62, 171.13, 163.55, 138.48, 137.22, 136.32, 134.68, 132.36, 131.55, 130.64, 129.93, 129.27, 129.18, 129.09, 128.56, 127.04, 126.62, 126.40, 126.08, 44.37, 18.49, 14.51。
[0028]
所得肟脂的高分辨质谱数据为:ultrflextof/tof (m/z)∶ c
24h17
no3s
+
,理论值401.1086,实测值401.1076 [m]
+
。
[0029]
测试例:准确称取三羟甲基丙烷三丙烯酸酯288mg(99 mol%)和本发明实施例1所得肟脂化合物3mg(1 mol%)并将其混合,将混合好的树脂超声10分钟以溶解、混合均匀。
[0030]
将上述混合物旋涂到载玻片表面,膜厚控制在0.5 mm左右。使用fusion uv光固化
系统(405 纳米光源,平均功率控制在1000 mw以下),照射350秒,测成膜后的铅笔硬度(gb/t 6739-1996,by型铅笔硬度计)为5h。
[0031]
将上述混合物滴涂到kbr盐片表面,膜厚控制在0.5 mm 左右,使用fusion uv光固化系统(405 纳米光源,平均功率控制在1000 mw以下),照射350秒,控制光源强度为25 mw/cm2,使用nicolet is50 ft-ir红外光谱仪对反应过程实时监测,测得成膜过程中双键转化率可达到75%,如图8所示。
[0032]
利用苯基-n-叔丁基硝酮为自由基捕获剂(与肟脂化合物加入比例为1∶1),以稳定光解时产生的自由基、延长其寿命,从而获得稳定的磁共振信号。由图9可知,照射产生的亚甲基自由基信号非常明显,其超精细裂分常数αn张量和αh张量分别为14.6 g和2.4 g,证实了此类自由基的存在。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1