一种核苷类似物及其盐的晶型、制备方法和应用与流程
1.本发明属于医药技术领域,具体涉及一种核苷类似物及其盐的晶型、制备方法和应用。
背景技术:
2.人类社会发展史,是跟病毒性传染病斗争的历史。近年来由于人类与动物之间复杂的相互作用以及地球生态系统的变化,新发和再发传染病流行加剧,对人类健康构成了严重威胁。这些病毒性传染病社会危害大,一直是公共卫生系统必须面对的巨大挑战。
3.病毒的遗传物质是dna或rna,其复制过程中需要大量的核苷酸,而核苷(酸)类似物可以模仿内源性核苷,在病毒合成dna或者rna过程中,与相应核苷竞争性结合病毒逆转录酶或rna聚合酶,终止病毒dna或rna链的延伸,从而抑制病毒复制。
4.申请公布号为cn 113735862a的发明专利申请公开了一种治疗病毒感染的核苷类化合物及其用途,该核苷类化合物及其前药,具有式i所述的化合物、其前药和/或其药学上可接受的盐。所述化合物及其组合物具有预防、缓解或治疗冠状病毒感染,或其同源变异病毒的复制或繁殖及其所产生的细胞病变效应的用途。
[0005][0006]
作为药物的同一化合物的不同晶型往往具有不同的稳定性。但是,到目前为止,对核苷类化合物的晶型研究少之又少。因此,本领域迫切需要开发物理性质稳定的、成药性好的核苷类似物的晶型用于治疗由病毒感染引起的相关疾病。
技术实现要素:
[0007]
本发明所要解决的技术问题是克服现有技术中的不足,提供一种核苷类似物及其盐的晶型、制备方法和应用,该核苷类似物及其盐的晶型物理性质稳定、成药性好,制备方法简单且易重复。
[0008]
为解决以上技术问题,本发明采取的技术方案是:
[0009]
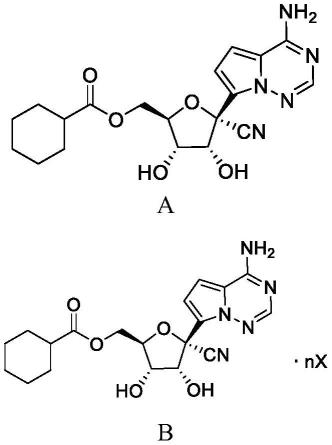
一种式a化合物,
[0010][0011]
式a化合物为结晶形式,结晶形式为晶型i,晶型i的x射线粉末衍射图谱在下列2θ值中的至少3处,优选为至少5处,更优选为至少7处具有特征峰: 9.85
°±
0.2
°
、17.42
°±
0.2
°
、17.75
°±
0.2
°
、18.72
°±
0.2
°
、19.39
°±
0.2
°
、23.90
°±ꢀ
0.2
°
和24.43
°±
0.2
°
。
[0012]
优选地,晶型i的x射线粉末衍射图谱还在下列2θ值中的至少3处,优选为至少5处,更优选为至少9处具有特征峰:12.94
°±
0.2
°
、13.18
°±
0.2
°
、15.62
°±
0.2
°
、19.64
°±
0.2
°
、21.32
°±
0.2
°
、23.57
°±
0.2
°
、28.20
°±
0.2
°
、30.57
°±
0.2
°
和31.18
°±
0.2
°
。
[0013]
优选地,晶型i的x射线粉末衍射图谱如图1所示。
[0014]
一种式a化合物,
[0015][0016]
式a化合物为无定形态。
[0017]
优选地,无定形态的x射线粉末衍射图谱如图3所示。
[0018]
为解决以上技术问题,本发明采取的又一技术方案是:
[0019]
一种如上所述的式a化合物结晶形式的制备方法,包括以下步骤:
[0020]
1)将式a化合物分散于溶剂s1中,加热、搅拌溶清,得到溶液s1’;
[0021]
2)将溶液s1’缓慢降温、搅拌析晶,滤过即得式a化合物的晶型i。
[0022]
优选地,在步骤1)中,式a化合物和溶剂s1的用量比为1g:20~100ml,优选为1g:50~80ml,
[0023]
搅拌时间为0.5~5小时,优选为0.5~1小时,
[0024]
加热温度为30~120℃,优选为50~90℃;
[0025]
在步骤2)中,析晶温度为-20~30℃,优选为-10℃~10℃,
[0026]
搅拌时间为0.5~5小时,优选为0.5~1小时。
[0027]
优选地,溶剂s1选自水、烃、醇、醚、酮、酯、腈及其均相混合物;
[0028]
烃选自正戊烷、正己烷、正庚烷、石油醚、二氯甲烷、氯仿、四氯化碳、 1,2-二氯乙烷、苯、甲苯、二甲苯、氯苯和二氯苯;
[0029]
醇选自甲醇、乙醇、正丙醇、异丙醇、正丁醇、乙二醇和丙二醇;
[0030]
醚选自乙醚、正丙醚、异丙醚、甲基叔丁基醚、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丙醚、乙二醇二甲醚、乙二醇二乙醚、丙二醇单甲醚、丙二醇单乙醚、丙二醇二甲醚、四氢呋喃、二氧六环、二甲氧基乙烷和二甘醇二甲醚;
[0031]
酮选自丙酮、丁酮和二乙基甲酮;
[0032]
酯选自甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、乙酸甲酯、乙酸乙酯、乙酸丙酯和乙酸丁酯;
[0033]
腈选自乙腈和丙腈。
[0034]
更优选地,溶剂s1选自甲醇。
[0035]
为解决以上技术问题,本发明采取的又一技术方案是:
[0036]
一种式a化合物的盐,其为式b化合物,
[0037][0038]
其中,x为马来酸、琥珀酸、柠檬酸、酒石酸、富马酸、甲酸、乙酸、丙酸、丙二酸、草酸、苯甲酸、邻苯二甲酸、甲磺酸、乙磺酸、苯磺酸、甲苯磺酸、萘磺酸、1,5-萘二磺酸、樟脑酸、樟脑磺酸、水杨酸、乙酰水杨酸、天门冬氨酸、谷氨酸、乳酸、葡萄糖酸、抗坏血酸、没食子酸、杏仁酸、苹果酸、山梨酸、三氟乙酸、牛磺酸、高牛磺酸、2-羟基乙磺酸、肉桂酸、粘酸、氯化氢、溴化氢、碘化氢、硫酸、硝酸、磷酸、高氯酸或其组合;
[0039]
x优选为马来酸、溴化氢、硝酸、甲磺酸、硫酸或其组合;
[0040]
n为0.5~2。
[0041]
优选地,盐为式b-1化合物,
[0042][0043]
其中,n为0.5~2。
[0044]
优选地,盐为式b-1’化合物,
[0045][0046]
优选地,盐为结晶形式,结晶形式为晶型ii,晶型ii的x射线粉末衍射图谱在下列2θ值中的至少3处,优选为至少5处,更优选为至少7处具有特征峰: 3.38
°±
0.2
°
、3.75
°±
0.2
°
、13.17
°±
0.2
°
、16.77
°±
0.2
°
、25.50
°±
0.2
°
、26.39
°±
0.2
°
和26.76
°±
0.2
°
。
[0047]
优选地,晶型ii的x射线粉末衍射图谱还在下列2θ值中的至少3处,优选为至少5处,更优选为至少9处具有特征峰:13.67
°±
0.2
°
、15.22
°±
0.2
°
、17.49
°±
0.2
°
、18.28
°±
0.2
°
、20.17
°±
0.2
°
、23.76
°±
0.2
°
、29.51
°±
0.2
°
、32.59
°±
0.2
°
和33.16
°±
0.2
°
。
[0048]
优选地,晶型ii的x射线粉末衍射图谱如图5所示。
[0049]
优选地,盐为无定形态。
[0050]
优选地,无定形态的x射线粉末衍射图谱如图7所示。
[0051]
为解决以上技术问题,本发明采取的又一技术方案是:
[0052]
一种如上所述的式a化合物的盐的结晶形式的制备方法,括以下步骤:
[0053]
1)将酸或者溶液s2’与溶液s1’混合,得到混合溶液,其中,溶液s2’是将酸溶于溶剂s2中得到的,溶液s1’是将式a化合物分散于溶剂s1中,加热、搅拌溶清得到的;
[0054]
2)将混合溶液在回流条件下搅拌并浓缩;
[0055]
3)将步骤2)中的产物与溶剂s3混合,在室温和/或加热条件下搅拌,降温析出固体,即得目标产物。
[0056]
优选地,在步骤1)中,溶液s2’中酸的含量为40wt%~50wt%,
[0057]
式a化合物与酸的摩尔比为1:0.9~1,
[0058]
式a化合物和溶剂s1的用量比为1g:20~100ml,优选为1g:50~80ml,
[0059]
搅拌时间为0.5~5小时,优选为0.5~1小时,
[0060]
加热温度为30~120℃,优选为50~90℃;
[0061]
在步骤2)中,搅拌时间为0.5~5小时,优选为0.5~1小时;
[0062]
在步骤3)中,式a化合物与溶剂s3的用量比为1g:10~50ml,优选为1 g:20~30ml,
[0063]
加热条件的温度为35~100℃,优选为50~60℃,
[0064]
搅拌的时间为0.5~5小时,优选为0.5~2小时,
[0065]
析晶温度为-20~30℃,优选为-10℃~10℃。
[0066]
优选地,溶剂s1、溶剂s2和溶剂s3各自独立地选自水、烃、醇、醚、酮、酯、腈及其均相混合物,且溶剂s1、s2和s3互溶;
[0067]
烃选自正戊烷、正己烷、正庚烷、石油醚、二氯甲烷、氯仿、四氯化碳、 1,2-二氯乙烷、苯、甲苯、二甲苯、氯苯和二氯苯;
[0068]
醇选自甲醇、乙醇、正丙醇、异丙醇、正丁醇、乙二醇和丙二醇;
[0069]
醚选自乙醚、正丙醚、异丙醚、甲基叔丁基醚、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丙醚、乙二醇二甲醚、乙二醇二乙醚、丙二醇单甲醚、丙二醇单乙醚、丙二醇二甲醚、四
氢呋喃、二氧六环、二甲氧基乙烷和二甘醇二甲醚;
[0070]
酮选自丙酮、丁酮和二乙基甲酮;
[0071]
酯选自甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、乙酸甲酯、乙酸乙酯、乙酸丙酯和乙酸丁酯;
[0072]
腈选自乙腈和丙腈。
[0073]
优选地,溶剂s1选自甲醇,溶剂s2选自水,溶剂s3选自甲基叔丁基醚。
[0074]
为解决以上技术问题,本发明采取的又一技术方案是:
[0075]
如上所述的式a化合物的结晶形式或如上所述的式a化合物的盐的结晶形式在制备治疗和/或缓解由病毒引起的疾病的药物中的应用,
[0076]
病毒优选为冠状病毒,
[0077]
冠状病毒优选为sars-cov-2。
[0078]
为解决以上技术问题,本发明采取的又一技术方案是:
[0079]
一种药物组合物,包含:
[0080]
1)如上所述的式a化合物的结晶形式或如上所述的式a化合物的盐的结晶形式;
[0081]
2)药学上可接受的辅料。
[0082]
优选地,药物组合物为口服制剂或非口服制剂;
[0083]
优选地,口服制剂选自片剂、胶囊剂、颗粒剂、散剂和糖浆剂;
[0084]
优选地,非口服制剂选自注射剂、粉针剂、喷剂和栓剂。
[0085]
优选地,辅料包括填充剂、崩解剂及润滑剂。
[0086]
优选地,填充剂选自乳糖、淀粉、改性淀粉、甘露醇、山梨醇、糊精衍生物、纤维素衍生物、碳酸钙、磷酸氢钙和硫酸钙中的一种或多种;其中,糊精衍生物选自糊精和/或麦芽糖糊精;纤维素衍生物选自微晶纤维素和/或纤维素。
[0087]
优选地,崩解剂选自交联羧甲基纤维素钠、交联聚维酮、淀粉、预胶化淀粉、羧甲基淀粉钠、羟丙基淀粉、微晶纤维素和低取代羟丙基纤维素中的一种或多种。
[0088]
优选地,润滑剂选自硬脂酸、硬脂酸钙、硬脂酸镁、氢化植物油、巴西棕榈蜡、滑石粉、聚乙二醇和硬脂富马酸钠中的一种或多种。
[0089]
由于以上技术方案的采用,本发明与现有技术相比具有如下优点:
[0090]
1、本发明核苷类似物具有晶型i的晶体形式,该晶型具有固体性状好、稳定性高等优点;
[0091]
2、本发明核苷类似物的酸加成盐(例如溴化氢盐)及其对应的晶型(例如晶型ii),溶解性较好;
[0092]
3、本发明核苷类似物及其盐的晶型物理性质稳定,易贮存,可用于制备治疗和/或缓解由病毒(特别是sars-cov-2)引起的相关疾病的药物;
[0093]
4、本发明的制备方法还具有产物晶型稳定、纯度高、易贮存、方法简单且易重复等优点。
附图说明
[0094]
图1为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯晶型i的xrpd图谱;
[0095]
图2为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯晶型i高温80℃、24h后的xrpd图谱;
[0096]
图3为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯无定形态的xrpd图谱;
[0097]
图4为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯晶型i的dsc图谱;
[0098]
图5为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐晶型ii的xrpd图谱;
[0099]
图6为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基氢溴酸盐晶型ii高温80℃、24h后的xrpd图谱;
[0100]
图7为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐无定形态的xrpd图谱;
[0101]
图8为((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐晶型ii的dsc图谱。
具体实施方式
[0102]
为使本发明的技术方案和有益效果能够更加明显易懂,下面通过结合附图和列举具体实施例的方式进行详细说明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规实验条件。
[0103]
[术语定义]
[0104]
核苷类似物
[0105]
核苷类似物是最重要的一类抗病毒药物。长期以来,在病毒性疾病的临床治疗中发挥了重要的作用。核苷类药物在生物体内可转化为相应的三磷酸形式,尤其是在病毒复制阶段,核苷三磷酸能“伪装”成底物,掺入到病毒的dna或 rna链中,从而抑制遗传物质的复制,发挥抗病毒作用。
[0106]
试剂和耗材说明
[0107]
在本发明的实施例及制备方法中涉及的甲醇、甲基叔丁基醚等试剂均为分析纯,由国药集团化学试剂有限公司提供,除非特别说明,所用试剂均未经过特别处理。高效液相色谱实验涉及的三乙胺、磷酸为色谱纯,由国药集团化学试剂有限公司提供。核磁共振谱在brucker 500hz和brucker 600hz核磁共振仪上测定。
[0108]
通用测试方法
[0109]
1、x射线粉末衍射(xrpd)测试方法:
[0110]
仪器:bruker d8 advance x射线多晶衍射仪;靶:cu-kα(40kv,40ma);样品到检测器距离:30cm;扫描类型:两轴联动;扫描步宽:0.02
°
;扫描范围: 3
°
~40
°
;扫描步径:0.1s。
[0111]
一般情况下,xrpd中的衍射角度(2θ值)可以在
±
0.2
°
的范围内产生误差,因此,本发明中涉及衍射角度的数值应当理解为也包含在约
±
0.2
°
的范围内的数值。所以,本发明不仅涵盖与具体xrpd图谱中的特征信号峰完全吻合的晶型,还涵盖与具体xrpd图谱中的特征信号峰存在
±
0.2
°
左右的误差的晶型。
[0112]
2、dsc测试方法:
[0113]
仪器:ta discovery dsc 2500差示扫描量热仪;温度范围:25-280℃;扫描速率:10℃/min;保护气体:氮气。
[0114]
3、稳定性测试方法:
[0115]
将供试品置于适宜的洁净容器中,分别放置于高温(80℃),高湿(25℃,相对湿度92.5%),光照(光照强度4500
±
500lx和90μw/cm2)条件下,放置7 天,于第7天取样,按稳定性考察项目进行检测。
[0116]
4、溶解度测试方法:
[0117]
取供试品约10mg,置于1.5ml的样品管中,加入去离子水1ml,置于超声仪中,于37℃超声30分钟;超声结束后,取上清液500μl,用乙腈稀释至合适浓度,作为供试品溶液,进行hplc分析,按外标法计算供试品在水(37℃) 中的溶解度。
[0118]
实施例1
[0119]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯晶型i
[0120][0121]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(400mg,1mmol)分散于甲醇中(32ml, 80v),升温至回流,搅拌30min产品彻底溶清,停止加热,自然冷却至室温,在0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色粉状固体,即为晶型 i(345mg,收率86%)。
[0122]1h nmr(400mhz,dmso-d6)δ7.92(m,3h),6.92(d,j=4.5hz,1h),6.81(d, j=4.5hz,1h),6.33(d,j=5.7hz,1h),5.37(d,j=5.9hz,1h),4.70(t,j=5.3hz, 1h),4.30(dd,j=12.1,2.8hz,1h),4.22(ddd,j=7.3,4.8,2.8hz,1h),4.14(dd,j =12.1,4.9hz,1h),3.96(q,j=5.8hz,1h),2.25(dt,j=10.7,5.9hz,1h),1.64(dd, j=39.6,31.8hz,5h),1.36
–
1.07(m,5h).
[0123]
13
c nmr(101mhz,dmso-d6)δ175.15,156.05,148.40,124.02,117.41, 117.04,110.67,101.21,81.65,79.41,74.53,70.58,62.99,42.60,28.98,28.89,25.73, 25.24,25.22.
[0124]
ms:m/z 402.21[m+1]
+
。
[0125]
差式扫描量热分析(dsc)结果显示,所得固体于227.1℃开始出现吸热峰,并于227.4℃时达到峰值,如图4所示。
[0126]
x射线粉末衍射(xrpd)结果显示,所得固体为晶体形式,对应于晶型i,如表1及图1所示。
[0127]
表1.式a化合物晶型i的xrpd数据表
[0128][0129]
实施例2
[0130]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯无定形态
[0131]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(100mg)加入到四氢呋喃(thf)(8ml) 中,回流搅拌溶清后,浓缩除去溶剂,油泵抽干,得到泡状固体(100mg)。x 射线粉末衍射(xrpd)结果显示,所得固体为无定形态,如图3所示。
[0132]
实施例3
[0133]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐晶型ii
[0134][0135]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基己基甲酸酯(300mg,0.75mmol)分散于甲醇中(24ml, 80v),升温至回流,搅拌30min后产品彻底溶清,加入48%氢溴酸溶液(60mg, 0.75mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约5ml),搅拌条件下缓慢加入甲基叔丁基醚(6ml,20v),50℃继续搅拌30min,冷却至 0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色粉状固体(256mg,收率71%)。
[0136]1h nmr(400mhz,dmso-d6)δ9.49(s,1h),8.91(s,1h),8.20(s,1h),7.33 (d,j=4.6hz,1h),6.97(d,j=4.7hz,1h),4.64(d,j=4.8hz,1h),4.35
–
4.21(m, 2h),4.20
–
4.06(m,1h),3.94(dd,j=7.0,4.6hz,1h),2.35
–
2.17(m,1h),1.87
–ꢀ
1.50(m,5h),1.35
–
1.12(m,5h).
[0137]
13
c nmr(101mhz,dmso-d6)δ173.01,140.76,127.95,127.61,116.93, 115.34,112.15,106.94,82.64,79.54,75.21,69.88,62.94,44.59,29.00,28.90,25.73, 25.25,25.22.
[0138]
ms:m/z 402.23[m+1]
+
。
[0139]
差式扫描量热分析(dsc)结果显示,所得固体于170.5℃开始出现吸热峰,并于178.4℃时达到峰值,如图8所示。
[0140]
x射线粉末衍射(xrpd)结果显示,所得固体为晶体形式,对应于晶型ii,如表2及图5所示。
[0141]
表2.式b-1’化合物晶型ii的xrpd数据表
[0142][0143]
实施例4
[0144]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐无定形态
[0145]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯氢溴酸盐(50mg)加入到thf(4ml)中,回流搅拌溶清后,浓缩除去溶剂,油泵抽干,得到泡状固体(50mg)。x射线粉末衍射(xrpd)结果显示,所得固体为无定形态,如图7所示。
[0146]
实施例5
[0147]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,
4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯马来酸盐
[0148][0149][0150]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(300mg,0.75mmol)分散于甲醇中(24ml, 80v),升温至回流,搅拌半小时后产品彻底溶清,加入马来酸(87mg,0.75 mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约5ml),搅拌条件下缓慢加入甲基叔丁基醚(6ml,20v),50℃继续搅拌30min,冷却至0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色粉状固体(256mg,收率71%)。
[0151]1h nmr(400mhz,dmso-d6)δ7.94(s,3h),6.93(d,j=4.5hz,1h),6.82(d, j=4.5hz,1h),6.34(s,1h),6.24(s,1h),5.38(s,1h),4.70(d,j=4.8hz,1h),4.30 (dd,j=12.1,2.8hz,1h),4.23(dq,j=7.2,2.9hz,1h),4.15(dd,j=12.1,4.9hz, 1h),3.96(dd,j=6.7,4.8hz,1h),2.24(ddt,j=10.8,7.2,3.6hz,1h),1.85
–
1.48 (m,5h),1.41
–
1.01(m,5h).
[0152]
ms:m/z 402.23[m+1]
+
。
[0153]
实施例6
[0154]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯甲磺酸盐
[0155][0156]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(300mg,0.75mmol)分散于甲醇中(24ml, 80v),升温至回流,搅拌半小时后产品彻底溶清,加入甲磺酸(72mg,0.75 mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约5ml),搅拌条件下缓慢加入甲基叔丁基醚(6ml,20v),50℃继续搅拌30min,冷却至0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色粘稠固体(288mg,收率77%)。
[0157]1h nmr(400mhz,dmso-d6)δ9.31(s,1h),8.83(s,1h),8.17(d,j=6.0hz, 1h),7.32
–
7.18(m,1h),6.95(d,j=4.6hz,1h),4.60(dd,j=30.3,4.9hz,1h), 4.33
–
4.21(m,2h),4.18
–
4.10(m,1h),3.94(t,j=5.8hz,1h),2.34(s,3h),2.26
ꢀ–
2.23(m,1h),1.87
–
1.51(m,5h),1.35
–
1.19(m,5h).
[0158]
ms:m/z 402.24[m+1]
+
。
[0159]
实施例7
[0160]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯硫酸盐
[0161][0162]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(100mg,0.25mmol)分散于甲醇中(8ml, 80v),升温至回流,搅拌半小时后产品彻底溶清,加入硫酸(25mg,0.25mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约1ml),搅拌条件下缓慢加入甲基叔丁基醚(2ml,20v),50℃继续搅拌30min,冷却至0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色泡状固体(98mg,收率79%)。
[0163]1h nmr(400mhz,dmso-d6)δ9.39(d,j=30.9hz,1h),8.87(s,1h),8.18 (d,j=6.0hz,1h),7.32
–
7.23(m,1h),7.00(dd,j=33.4,4.6hz,1h),4.57(d,j=4.9hz,1h),4.31
–
4.22(m,2h),3.66
–
3.60(m,1h),3.56
–
3.49(m,1h),2.30
–ꢀ
2.11(m,1h),1.95
–
1.43(m,5h),1.44
–
0.98(m,5h).
[0164]
ms:m/z 402.21[m+1]
+
。
[0165]
实施例8
[0166]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯半硫酸盐
[0167][0168]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(100mg,0.25mmol)分散于甲醇中(8ml, 80v),升温至回流,搅拌半小时后产品彻底溶清,加入硫酸(13mg,0.13mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约1ml),搅拌条件下缓慢加入甲基叔丁基醚(2ml,20v),50℃继续搅拌30min,冷却至0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色泡状固体(98mg,收率88%)。
[0169]1h nmr(400mhz,dmso-d6)δ8.80(s,1h),8.45(s,1h),8.08(d,j=5.7hz, 1h),7.13(d,j=4.5hz,1h),6.90(d,j=4.6hz,1h),4.66(d,j=4.8hz,1h),4.33
ꢀ–
4.20(m,2h),4.15
(dd,j=12.0,4.7hz,1h),3.95(dd,j=6.6,4.8hz,1h),2.30
–ꢀ
2.20(m,1h),1.83
–
1.51(m,5h),1.35
–
1.14(m,5h).
[0170]
ms:m/z 402.23[m+1]
+
。
[0171]
实施例9
[0172]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯磷酸盐
[0173][0174]
将((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(100mg,0.25mmol)分散于甲醇中(8ml, 80v),升温至回流,搅拌半小时后产品彻底溶清,加入磷酸(25mg,0.25mmol),继续搅拌30min,待其冷却至室温,浓缩至小体积(约1ml),搅拌条件下缓慢加入甲基叔丁基醚(2ml,20v),50℃继续搅拌30min,冷却至0~10℃继续搅拌30min,过滤,滤饼油泵拉干,得白色泡状固体(85mg,收率69%)。
[0175]1h nmr(400mhz,dmso-d6)δ7.92(s,3h),6.91(dd,j=9.6,5.4hz,1h), 6.81(d,j=4.5hz,1h),4.70(d,j=4.8hz,1h),4.30(dd,j=12.1,2.8hz,1h), 4.22(dt,j=7.2,3.8hz,2h),4.16(d,j=4.9hz,1h),2.27
–
2.18(m,1h),1.82
–ꢀ
1.49(m,5h),1.35
–
1.11(m,5h).
[0176]
ms:m/z 402.34[m+1]
+
。
[0177]
实施例10
[0178]
制备((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯盐酸盐
[0179][0180]
参照实施例2-7的方法,化合物((2r,3s,4r,5r)-5-(4-氨基吡咯并[2,1-f][1,2,4] 三嗪-7-基)-5-氰基-3,4-二羟基四氢呋喃-2-基)甲基环己基甲酸酯(300mg,0.75 mmol)与盐酸(76mg,0.75mmol)成盐,析出的固体粘壁,无法过滤。
[0181]
实施例11
[0182]
化合物a及其酸加成盐的性状、稳定性比较。
[0183]
(1)化合物a及其酸加盐的性状、制备情况比较
[0184]
选取化合物a及其不同的盐(通过实施例1-8制备),比较其性状及制备难易程度,结果见表3。
[0185]
表3.化合物a及其盐的性状及制备情况表
[0186]
化合物性状制备情况化合物a晶型i白色粉状固体操作简单,固体自然沉降,易过滤氢溴酸盐(b-1’)白色粉状固体操作简单,固体自然沉降,易过滤马来酸盐(b-2’)白色粉状固体操作简单,固体自然沉降,易过滤甲磺酸盐(b-3’)白色粘稠固体固体粘壁,易吸潮,难过滤硫酸盐(b-4’)泡状固体固体不能自然沉降,易过滤,略吸潮半硫酸盐(b-5’)泡状固体固体不能自然沉降,易过滤,略吸潮磷酸盐(b-6’)固体性状差固体粘壁,难过滤,易吸潮盐酸盐(b-7’)未得到固体/
[0187]
由上表可见,化合物a的氢溴酸和马来酸加成盐形式性状良好,且更容易制备。
[0188]
(2)化合物a加成盐的稳定性比较
[0189]
选取化合物a不同的盐(通过实施例2-7制备),比较其稳定性,结果见表 4。
[0190]
表4.化合物a加成盐的稳定性情况表
[0191][0192][0193]
由上表可见,化合物a的氢溴酸盐和马来酸盐在高温和光照条件下均很稳定,高湿条件下略吸潮,而化合物a甲磺酸盐、硫酸盐、半硫酸盐和磷酸盐的稳定性相对较差。
[0194]
(3)化合物a及其盐的溶解性比较
[0195]
选取化合物a及其不同的盐(实施例1、2和3制备),比较其溶解度。
[0196]
称取适量样品,置于玻璃样试管中,逐步递增加入选定的溶媒,观察澄清情况。测定了化合物a及其不同的盐在去离子水中的溶解度,结果见表5。
[0197]
表5.化合物a及其盐在去离子水中的溶解度
[0198][0199]
由上表可见,化合物a的水溶性明显小于化合物a的盐,其中氢溴酸盐水溶性最好,而水溶性大小在药物制剂、口服生物用度等方面具有举足轻重的影响。因此,将化合物a成氢溴酸盐更有利于制成药物制剂,拓宽其药物用途。
[0200]
实施例12
[0201]
化合物a的晶型i的稳定性考察
[0202]
如图1和图2所示,在高温80℃,24h后,晶型i没有变化,稳定性好。
[0203]
实施例13
[0204]
化合物a氢溴酸盐的晶型ii的稳定性考察
[0205]
如图5和图6所示,在高温80℃,24h后,晶型ii基本不变。
[0206]
本发明对具有抗病毒活性(特别是抗sars-cov-2活性)的核苷类似物的晶型,其酸加成盐、基于该酸加成盐得到的盐型晶体进行了合成、分离,以及相关的理化性质研究。
[0207]
经研究发现了该核苷类似物具有晶型i的晶体形式,该晶型具有固体性状好、稳定性高等优点。另外,发现了该核苷类似物的酸加成盐(例如溴化氢盐) 及其对应的晶型(例如晶型ii),溶解性较好。核苷类似物及其盐的晶型可用于制备治疗和/或缓解由病毒(特别是sars-cov-2)引起的相关疾病的药物。除此之外,本发明中的制备方法还具有产物晶型稳定、纯度高、易贮存、方法简单且易重复等优点。
[0208]
应当理解,以上实施例均为示例性的,不用于包含权利要求所包含的所有可能的实施方式。在不脱离本公开的范围的情况下,还可以在以上实施例的基础上做出各种变形和改变。同样的,也可以对以上实施例的各个技术特征进行任意组合,以形成可能没有被明确描述的本发明的另外的实施例。因此,上述实施例仅表达了本发明的几种实施方式,不对本发明专利的保护范围进行限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1