含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物及其制备方法和应用
1.本发明属于化学合成和药物应用技术领域,具体涉及一种含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物及其制备方法和应用。
背景技术:
2.化学农药以其高效、高速、低成本、简便的特点,在农作物种植过程中有害生物综合治理方面有着举足轻重的作用,其种类繁多,应用广泛,是保障农业生产、农作物品质的有效手段。随着农产品需求的增加和“环境友好”观念的深入,高效、广谱、低毒化学农药的开发是新农药创制的未来趋势,其中杂环类有机农药为近年来发展最为迅速的一类农药。1,2,4-噁二唑类杂环化合物具有广泛的生物活性,如杀虫、杀菌及除草等。因此,1,2,4-噁二唑类化合物的分子设计、合成与生物活性研究是当今绿色农药创制的热点。
技术实现要素:
3.针对上述问题,本发明的目的在于提供一种含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物及其制备方法和用。
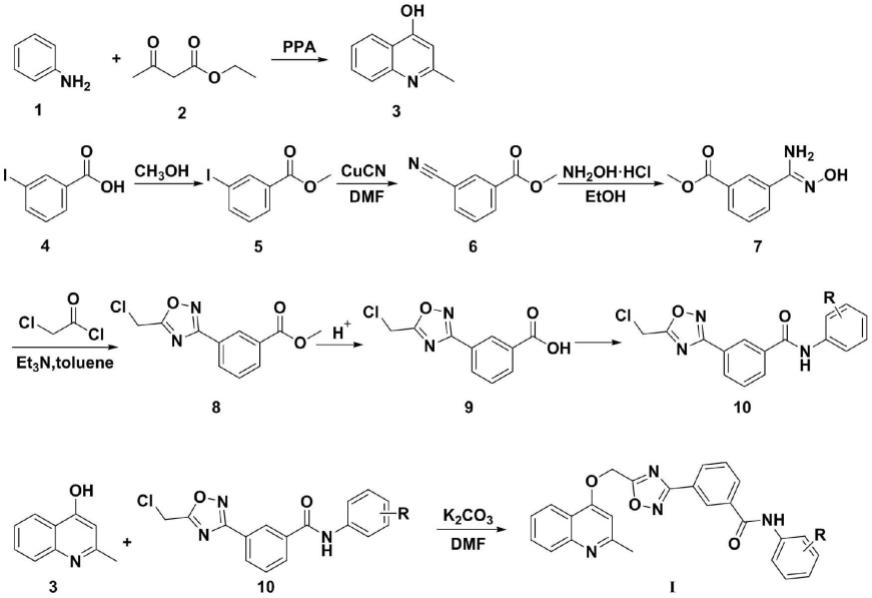
4.本发明公开了所述的含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物,含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物,其结构式如式(i)所示:
[0005][0006]
式(i)中,r为取代基,苯环上的h被取代基r单取代、多取代或不被取代,单取代或多取代的取代基r各自独立地选自烷基、氟取代烷基或卤素。
[0007]
所述的含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物,其特征在于取代基r各自独立为甲基、三氟甲基、f、cl、br或i;r优选为h、2-甲基、3-甲基、4-甲基、4-叔丁基、3-三氟甲基、2-氟、3-氟、4-氟、4-氯、4-溴、4-碘、2,4-二甲基、2,6-二甲基、3-氯-2-甲基、3,4-二氯或2,4-二氟。
[0008]
本发明还公开了含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物的制备方法,包括如下步骤:
[0009]
1)以多聚磷酸(ppa)为催化剂和溶剂,将式(1)所示的苯胺和式(2)所示的乙酰乙酸乙酯进行环化反应,经过滤,洗涤和干燥后得到式(3)所示的2-甲基-4-羟基喹啉;
[0010]
2)以浓硫酸为催化剂,将式(4)所示的3-碘苯甲酸和甲醇进行酯化,生成式(5)所示的3-碘苯甲酸甲酯;
[0011]
3)以dmf为溶剂,将步骤2)所得的3-碘苯甲酸甲酯与氰化亚铜在l-脯氨酸催化下,
反应生成如式(6)所示的3-氰基苯甲酸甲酯;
[0012]
4)以乙醇为溶剂,向步骤3)所得的3-氰基苯甲酸甲酯中缓慢加入盐酸羟胺和三乙胺,生成式(7)所示的中间体7;
[0013]
5)以甲苯为溶剂,加入三乙胺,将步骤4)所得的中间体7与氯乙酰氯进行环合反应,生成式(8)所示的中间体8,在酸性条件下水解,生成式(9)所示的中间体9。
[0014]
6)以thf为溶剂,将步骤5)所得的中间体9与取代苯胺进行缩合反应,生成式(10)所示中间体10;
[0015]
7)以dmf为溶剂,将步骤1)所得的2-甲基-4-羟基喹啉和步骤6)所得的中间体10进行醚化反应,得如式(i)所示的目标产物含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物;
[0016]
其反应过程如下:
[0017][0018]
式(i)中,苯环上的h被取代基r单取代、多取代或不可取代,单取代或多取代的取代基r各自独立地选自烷基、氟取代烷基或卤素。
[0019]
进一步地,步骤1)中苯胺和乙酰乙酸乙酯的摩尔比为1:1-2。
[0020]
进一步地,步骤3)中3-碘苯甲酸甲酯,氰化亚铜和l-脯氨酸的摩尔比为1:1.3-1.7:1。
[0021]
进一步地,步骤4)中3-氰基苯甲酸甲酯,盐酸羟胺与三乙胺的摩尔比为1:1.5-1.8:1.4-1.7。
[0022]
进一步地,步骤5)中三乙胺,中间体7和氯乙酰氯的摩尔比为2.3-2.6:1:1.0-1.4。中间体9,取代苯胺与三乙胺的摩尔比为1:1.2-1.5:2.5-3。
[0023]
进一步地,步骤7)中2-甲基-4-羟基喹啉和中间体10的摩尔比为1:1-1.5。
[0024]
本发明还研究了所得到的含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物在制备杀菌剂中的应用。
[0025]
本发明的制备方法简单、操作方便,所得产物的结构经核磁氢谱、碳谱和高分辨质
谱进行了确认,并对所得的17个目标产物进行了杀菌活性测试,结果表明:在50ppm浓度下,通过本发明的制备方法得到的目标产物表现出良好抑菌活性,其中对油菜菌核病菌,化合物ia的抑制率达86.1%。
具体实施方式
[0026]
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
[0027]
实施例1式(3)所示2-甲基-4-羟基喹啉的制备
[0028]
在250ml三口烧瓶中加入50.69g多聚磷酸(ppa),然后升温至90℃保持0.5h,搅拌下缓慢加入9.31g(0.10mol)苯胺和13.14g(0.10mol)乙酰乙酸乙酯,继续升温至150℃反应5h,tlc监测反应结束。待反应液冷却至室温,加入100ml水搅拌析出灰色固体,抽滤,依次用用石油醚和饱和碳酸氢钠水溶液洗涤,烘干得产物14.20g,收率89.4%。
[0029]
实施例2式(5)所示3-碘苯甲酸甲酯的制备
[0030]
在100ml三口烧瓶中加入3-碘苯甲酸2.53g(0.01mol)、50ml甲醇,滴加0.50ml浓硫酸,升温至回流反应,tlc跟踪监测反应进程,约8h结束。待反应液冷却至室温,旋蒸脱溶,加入50ml乙酸乙酯,用饱和碳酸钠水溶液调节体系ph值至7~8,分液取有机相,用无水硫酸钠干燥脱溶得2.39g米白色固体,收率90.4%。
[0031]
实施例3式(6)所示3-氰基苯甲酸甲酯的制备
[0032]
在100ml的三口烧瓶中加入实施例2制备得到的3-碘苯甲酸甲酯0.34g(1.30mmol)、氰化亚铜0.18g(2.00mmol)、l-脯氨酸0.15g(1.30mmol)和15ml dmf,待溶解后,升温至75℃,反应2h。继续升温至100℃反应,tlc跟踪监测反应,约6h结束。待反应液冷却至室温,用硅藻土抽滤除去滤渣,滤液加入100ml水和100ml乙酸乙酯萃取,有机相水洗(50ml
×
3),然后用无水硫酸镁干燥,旋蒸脱除乙酸乙酯,得0.17g黄色固体,收率81.9%。
[0033]
实施例4式(7)所示中间体7的制备
[0034]
在100ml三口烧瓶中加入实施例3制备得到的3-氰基苯甲酸甲酯1.16g(7.20mmol),45ml无水乙醇。在室温下,开启机械搅拌,随后缓慢加入盐酸羟胺0.75g(10.79mmol)、三乙胺1.10g(10.87mmol)开始反应,tlc跟踪监测反应进程,约3h结束。旋蒸脱溶,并将残余物溶于50ml乙酸乙酯和50ml饱和氯化钠水溶液中,分液取有机层,加入无水硫酸钠干燥,旋蒸脱溶得到淡黄色固体1.26g,收率90.3%。
[0035]
实施例5式(8)所示中间体8的制备。
[0036]
在250ml三口烧瓶中加入实施例4制备得到的中间体70.97g(5.00mmol),三乙胺1.20g(12.00mmol),100ml无水甲苯,控温0℃,搅拌2h后,逐滴加入0.58g(5.20mmol)氯乙酰氯,0℃下继续搅拌反应3h,然后升温至回流反应,tlc监测反应进程,约2h结束。待反应液冷却至室温,用150ml蒸馏水进行水洗,然后用饱和氯化钠水溶液洗涤,有机相加入无水硫酸钠干燥,旋蒸脱溶,烘干得到产物0.93g,收率73.8%。
[0037]
实施例6式(9)所示中间体9的制备。
[0038]
在250ml的三口烧瓶中依次加入实施例5制备得到的5.00g(0.02mol)中间体8,30ml乙酸,30ml盐酸,升温至70℃反应3h,tlc监测反应结束。待反应液冷却至室温,析出大量白色固体,抽滤,水洗烘干得产物4.45g,收率93.6%,m.p.179~182℃;1h nmr(400mhz,dmso-d6)δ:13.34(s,1h),8.53(s,1h),8.23(d,j=8.8hz,1h),8.14(d,j=7.8hz,1h),7.71
(t,j=7.8hz,1h),5.19(s,2h).
[0039]
实施例7式(10)所示中间体10a-10q的制备。
[0040]
在50ml的三口烧瓶中加入实施例6制备得到的0.24g(1.00mmol)中间体9,5ml二氯亚砜,加热搅拌回流反应3h。旋蒸脱除二氯亚砜,加入30ml thf,冰浴条件下逐滴加入1.20mmol取代苯胺(苯环上的h被取代基r取代或不取代,r为2-甲基、3-甲基、4-甲基、4-叔丁基、3-三氟甲基、2-氟、3-氟、4-氟、4-氯、4-溴、4-碘、2,4-二甲基、2,6-二甲基、3-氯-2-甲基、3,4-二氯或2,4-二氟),2.50mmol三乙胺,1ml thf的混合溶液,搅拌过夜经柱层析分离纯化得中间体10a-10q。具体数据见表1,表2,表3,表4。
[0041]
表1中间体10a-10q理化数据
[0042]
中间体取代基团r外观收率%10ah黄色固体79.410b2-甲基黄色固体77.510c3-甲基白色固体69.710d4-甲基白色固体73.810e4-叔丁基白色固体75.710f3-三氟甲基黄色固体66.410g2-氟黄色固体75.910h3-氟黄色固体78.610i4-氟黄色固体67.110j4-氯黄色固体79.310k4-溴黄色固体73.410l4-碘黄色固体68.810m2,4-二甲基白色固体69.510n2,6-二甲基白色固体74.410o3-氯-2甲基白色固体63.710p3,4-二氯黄色固体64.910q2,4-二氟黄色固体63.7
[0043]
表2中间体10a-10q氢谱数据
[0044]
[0045][0046]
表3中间体10a-10q碳谱数据
[0047]
[0048][0049]
表4中间体10a-10q高分辨质谱数据
[0050]
[0051][0052]
实施例8式(i)所示目标化合物ia-iq的合成。
[0053]
在100ml的单口烧瓶中加入实施例1得到的(1.00mmol)2-甲基4-羟基喹啉,和实施例7得到的(1.00mmol)中间体10,0.35g k2co3,10ml dmf,升温至60℃反应5h,tlc监测反应结束。待反应液冷却至室温,将其倒入100ml水中,并用乙酸乙酯萃取,无水硫酸镁干燥脱溶得粗产物,经快速柱层析分离纯化得目标化合物ia-iq。具体数据见表5,表6,表7和表8。
[0054]
表5含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物理化数据
[0055]
目标化合物取代基团r外观收率%iah黄色固体63.1ib2-甲基黄色固体61.7ic3-甲基黄色固体56.4id4-甲基黄色固体67.4ie4-叔丁基黄色固体53.8if3-三氟甲基黄色固体56.7ig2-氟黄色固体58.1ih3-氟黄色固体71.4ii4-氟黄色固体66.7ij4-氯白色固体58.9ik4-溴黄色固体54.3il4-碘黄色固体57.4im2,4-二甲基黄色固体65.3in2,6-二甲基黄色固体67.2io3-氯-2甲基黄色固体56.2ip3,4-二氯黄色固体51.7iq2,4-二氟黄色固体47.6
[0056]
表6含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物氢谱数据
[0057]
[0058][0059]
[0060]
表7含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物碳谱数据
[0061][0062][0063]
表8含喹啉环1,2,4-噁二唑取代苯甲酰胺类化合物高分辨质谱数据
[0064][0065]
实施例9杀菌活性测试。
[0066]
1测试样品
[0067]
实验对象:番茄早疫病菌(alternaria solani)、小麦赤霉病菌(gibberellazeae)、水稻稻瘟病菌(pyriculariaoryae)、辣椒疫霉病菌(phytophthora capsici)、油菜菌核病菌(sclerotinia sclerotiorum)、黄瓜灰霉病菌(botrytis cinerea)、水稻纹枯病菌(riziocotiniasolani)、黄瓜枯萎病菌(fusarium oxysporum)、花生褐斑病菌(cercosporaarachidicola)以及苹果轮纹病菌(physalosporapiricola)。
[0068]
实验处理:将各待测化合物用dmso溶解制成1%ec母液备用。试验采用含毒培养基法,评价供试化合物在50ppm的剂量下对10种试验靶标的室内杀菌活性,活性测试结果如表9所示;
[0069]
表9化合物ia~iq的杀菌活性(50mg/l)
[0070][0071]
目标化合物ia-iq的杀菌活性测试结果见表9。从表9可以看出:在50mg/l测试浓度下,目标化合物iva对油菜菌核病菌显示出良好的抑制活性。其中,化合物ia、ii、ij、ik和il的抑制率达80.6%~86.1%,优于对照药苯氧喹啉(77.8%)。化合物ic、id、if、ih和im的抑制率分别为75.0%、77.8%、77.8%、75.0%和77.8%,与苯氧喹啉药效相当。此外,化合物ig、io和ip也有中等以上的抑制活性(61.1%~69.4%)。对水稻稻瘟病,化合物ib、ij、ik和ip显示出一定的活性,抑制率均为46.7%,与苯氧喹啉药效相当(46.7%)。对于其他病菌,化合物ia~iq与苯氧喹啉一样,药效一般。
[0072]
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式,本发明的保护范围也仅仅于本领域技术人员根据本发明构思所能够想到的等同技术手段。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1