基因编辑系统及其在构建TNIP1基因突变的银屑病模型猪核移植供体细胞中的应用的制作方法
基因编辑系统及其在构建tnip1基因突变的银屑病模型猪核移植供体细胞中的应用
技术领域
1.本发明属于生物技术领域,具体属于基因编辑技术领域,更具体涉及基因编辑系统及其在构建tnip1基因突变的银屑病模型猪核移植供体细胞中的应用。
背景技术:
2.银屑病,又称“白疕”或“牛皮癣”,是一种慢性皮肤疾病,该病的典型临床表现为皮肤出现鳞屑性红斑,严重者可引起致残性银屑病性关节炎,该病严重影响患者的生活,对患者及其家庭造成巨大经济负担。在欧洲和北美人群中,银屑病的发病率为1%-2%,在中国人群中,银屑病患病率虽然仅为0.47%,但由于国内人口基数大,仍有着相对庞大的患病人群。目前尚未有根治银屑病的方法。
3.家族聚集性研究证实银屑病能够遗传,且为多基因遗传性疾病。tnfα诱导蛋白3相互作用蛋白1(tnfα-induced protein 3-interacting protein 1,tnip1)基因,为中国汉族和欧美人群所共有的一个银屑病易感基因。tnipi基因编码的tnip1蛋白,与tnip2、tnip3同属于tnips家族,该家族因能与泛素编辑蛋白a20结合、并抑制核转录因子(nuclear factor-κb,nf-κb)的转录活性而得名,并因此获得另一个命名abins(a20-binding inhibitor of nf-κb)。此外,tnip1还可不依赖于a20和nf-κb发挥作用,细胞外信号调节酶1/2(erk 1/2)、视黄酸受体-α/γ(rar-α/γ)、过氧化物酶体增殖物激活受体(ppar)和ccaat/增强子结合蛋白β(c/ebpβ)等均被证实为tnip1的靶目标,这些分子被tnip1激活后可通过多条信号通路发挥不同的生物学作用。
4.研究银屑病的发生发展机制及研发相应的药物均需要在动物模型的基础上进行,目前常用的动物模型为小鼠模型,然而小鼠不论从体型、器官大小、生理、病理等方面都与人相差巨大,不能真实地模拟人类正常的生理、病理状态。猪作为大动物,其体型大小和生理功能与人类近似,易于大规模繁殖饲养,而且在伦理道德及动物保护等方面要求较低,是理想的人类疾病模型动物。
5.基因编辑是近年来不断取得重大发展的一种生物技术,其包括从基于同源重组的基因编辑到基于核酸酶的zfn、talen、crispr/cas9等编辑技术,其中crispr/cas9技术是当前最先进的基因编辑技术。目前,基因编辑技术被越来越多地应用到动物模型的制作上。
技术实现要素:
6.本发明的目的是提供基因编辑系统及其在构建tnip1基因突变的银屑病模型猪核移植供体细胞中的应用。
7.本发明提供了一种试剂盒,包括tnip1-grna2、tnip1-grna3和ncn蛋白。
8.本发明还提供了一种试剂盒,包括tnip1-grna2、tnip1-grna3和proncn蛋白。
9.本发明还提供了一种试剂盒,包括tnip1-grna2、tnip1-grna3和特异质粒。
10.以上任一所述试剂盒还包括猪细胞。
11.本发明提供了tnip1-grna2、tnip1-grna3和ncn蛋白在制备试剂盒中的应用。
12.本发明还提供了tnip1-grna2、tnip1-grna3和proncn蛋白在制备试剂盒中的应用。
13.本发明还提供了tnip1-grna2、tnip1-grna3和特异质粒在制备试剂盒中的应用。
14.以上任一所述试剂盒的用途为如下(a)或(b)或(c):(a)制备重猪组细胞;(b)制备银屑病模型猪;(c)制备银屑病细胞模型或银屑病组织模型或银屑病器官模型。
15.本发明提供了一种制备重组猪细胞的方法,包括如下步骤:将tnip1-grna2、tnip1-grna3和ncn蛋白共转染猪细胞,得到重组猪细胞。
16.所述共转染具体采用电击转染的方式。
17.电击转染的参数设置具体可为:1450v、10ms、3pulse。
18.所述共转染具体可采用哺乳动物核转染试剂盒(neon kit,thermofisher)与neon tm transfection system电转仪。
19.tnip1-grna2、tnip1-grna3和ncn蛋白的配比依次为:0.8-1.2μg tnip1-grna2:0.8-1.2μg tnip1-grna3:3-5μg ncn蛋白。
20.tnip1-grna2、tnip1-grna3和ncn蛋白的配比依次为:1μg tnip1-grna2:1μgtnip1-grna3:4μg ncn蛋白。
21.猪细胞、tnip1-grna2、tnip1-grna3和ncn蛋白的配比依次为:10万个猪细胞:0.8-1.2μg tnip1-grna2:0.8-1.2μg tnip1-grna3:3-5μg ncn蛋白。
22.猪细胞、tnip1-grna2、tnip1-grna3和ncn蛋白的配比依次为:10万个猪细胞:1μgtnip1-grna2:1μg tnip1-grna3:4μg ncn蛋白。
23.以上任一所述tnip1-grna2为sgrna,其靶序列结合区如seq id no:16中第3-22位核苷酸所示。
24.具体的,所述tnip1-grna2如seq id no:16所示。
25.具体的,所述tnip1-grna2如seq id no:11所示。
26.以上任一所述tnip1-grna3为sgrna,其靶序列结合区如seq id no:17中第3-22位核苷酸所示。
27.具体的,所述tnip1-grna3如seq id no:17所示。
28.具体的,所述tnip1-grna3如seq id no:12所示。
29.以上任一所述ncn蛋白为cas9蛋白或具有cas9蛋白的融合蛋白。
30.具体的,所述ncn蛋白如seq id no:3所示。
31.以上任一所述猪细胞为猪成纤维细胞。
32.以上任一所述猪细胞为猪原代成纤维细胞。
33.以上任一所述猪细胞为获自初生猪的猪原代成纤维细胞。
34.所述ncn蛋白的制备方法包括如下步骤:
35.(1)将质粒pkg-ge4导入大肠杆菌bl21(de3),得到重组菌;
36.(2)采用液体培养基30℃培养所述重组菌,然后加入iptg并进行25℃诱导培养,然后收集菌体;
37.(3)将收集的菌体进行菌体破碎,收集粗蛋白溶液;
38.(4)采用亲和层析从所述粗蛋白溶液中纯化具有his6标签的融合蛋白;
39.(5)采用具有his6标签的肠激酶酶切具有his6标签的融合蛋白,然后采用ni-nta树脂去除具有his6标签的蛋白,得到纯化的ncn蛋白;
40.质粒pkg-ge4中具有seq id no:1中第5209-9852位核苷酸所示的融合基因。
41.所述ncn蛋白的制备方法具体包括如下步骤:
42.(1)将质粒pkg-ge4导入大肠杆菌bl21(de3),得到重组菌。
43.(2)将步骤(1)得到的重组菌接种至含氨苄青霉素的液体lb培养基,振荡培养;
44.(3)将步骤(2)得到的菌液接种至液体lb培养基,30℃、230rpm振荡培养至od
600nm
值=1.0,然后加入iptg并使其在体系中的浓度为0.5mm,然后25℃、230rpm振荡培养12小时,然后离心收集菌体;
45.(4)取步骤(3)得到的菌体,用pbs缓冲液洗涤;
46.(5)取步骤(4)得到的菌体,加入粗提缓冲液并悬浮菌体,然后进行菌体破碎,然后离心收集上清液,采用0.22μm孔径滤膜过滤,收集滤液;
47.(6)采用亲和层析从步骤(5)得到的滤液中纯化具有his6标签的融合蛋白(seq id no:2所示的融合蛋白);
48.(7)取步骤(6)收集的过柱后溶液,使用超滤管浓缩,然后用25mm tris-hcl(ph8.0)稀释;
49.(8)将具有his6标签的重组牛肠激酶加入到步骤(7)得到的溶液中,酶切;
50.(9)将完成步骤(8)的溶液与ni-nta树脂混匀,孵育,然后离心收集上清液;
51.(10)取步骤(9)得到的上清液,使用超滤管浓缩,然后加入酶贮存液中,即为ncn蛋白溶液。
52.采用亲和层析从步骤(5)得到的滤液中纯化具有his6标签的融合蛋白的具体方法如下:
53.首先采用5个柱体积的平衡液平衡ni-nta琼脂糖柱(流速为1ml/min);然后上样50ml步骤(5)得到的滤液(流速为0.5-1ml/min);然后用5个柱体积的平衡液洗涤柱子(流速为1ml/min);然后用5个柱体积的缓冲液洗涤柱子(流速为1ml/min),以去除杂蛋白;然后用10个柱体积的洗脱液以0.5-1ml/min的流速洗脱,收集过柱后溶液(90-100ml)。
54.以上任一所述proncn蛋白自上游至下游依次包括如下元件:信号肽、分子伴侣蛋白、蛋白标签、蛋白酶酶切位点、核定位信号、cas9蛋白、核定位信号。
55.所述信号肽的功能为促进蛋白分泌表达。所述信号肽可选自大肠杆菌碱性磷酸酶(phoa)信号肽、金黄色葡萄球菌蛋白a信号肽、大肠杆菌外膜蛋白(ompa)信号肽或任何其他原核基因的信号肽,优选为碱性磷酸酶信号肽(phoa signal peptide)。碱性磷酸酶信号肽用来引导目的蛋白分泌表达至细菌周质腔中,从而与细菌胞内蛋白分离,且分泌到细菌周质腔中的目的蛋白为可溶性表达,可被细菌周质腔中的信号肽酶裂解。
56.所述分子伴侣蛋白的功能为增加蛋白的可溶性。所述分子伴侣可为任何帮助形成二硫键的蛋白,优选为硫氧还原蛋白(trxa蛋白)。硫氧还原蛋白,其能作为分子伴侣帮助所共表达的目的蛋白(例如cas9蛋白)形成二硫键,提高蛋白的稳定性、折叠的正确性,增加目的蛋白的溶解性及活性。
57.所述蛋白标签的功能为用于蛋白纯化。所述标签可为his标签(his-tag,his6蛋白标签)、gst标签、flag标签、ha标签、c-myc标签或其他任何蛋白标签,进一步优选为his标
签。his标签能与ni柱结合,可以通过一步法ni柱亲和层析纯化目的蛋白,可极大地简化目的蛋白的纯化流程。
58.所述蛋白酶酶切位点的功能为纯化后用于切除非功能区段,以释放天然形式cas9蛋白。所述蛋白酶可选自肠激酶(enterokinase)、因子xa(factor xa)、凝血酶(thrombin)、tev蛋白酶(tev protease)、hrv 3c蛋白酶(hrv 3c protease)、welqut蛋白酶或任何其他内切蛋白酶,进一步优选为肠激酶。ek为肠激酶酶切位点,便于使用肠激酶切除所融合的trxa-his区段,得到天然形式的cas9蛋白。本技术使用带his标签的商品肠激酶酶切融合蛋白后,可通过一次亲和层析除去trxa-his区段及带his标签的肠激酶,得到天然形式的cas9蛋白,避免了多次纯化透析对目的蛋白的伤害和损耗。
59.所述核定位信号可为任何核定位信号,优选为sv40核定位信号和/或nucleoplasmin核定位信号。nls为核定位信号,在cas9的n端及c端分别设计了一个nls位点,使cas9能更有效地进入细胞核进行基因编辑。
60.所述cas9蛋白可为sacas9或spcas9,优选为spcas9蛋白。
61.proncn蛋白具体如seq id no:2所示。
62.以上任一所述特异质粒自上游至下游依次包括如下元件:启动子、操纵子、核糖体结合位点、proncn蛋白的编码基因、终止子。
63.所述启动子具体可为t7启动子。t7启动子为原核表达强启动子,能高效驱动外源基因的表达。
64.所述操纵子具体可为lac操纵子。lac操纵子为乳糖诱导表达的调控元件,可在细菌生长至一定数量后,再用iptg在低温下诱导目的蛋白的表达,可避免目的蛋白过早表达对宿主菌生长的影响,低温下诱导表达也显著提高所表达的目的蛋白的可溶性。
65.所述核糖体结合位点是蛋白翻译时的核糖体结合位点,对蛋白质的翻译是必要的。
66.所述终止子具体可为t7终止子。t7终止子可在目的基因的末端有效终止基因转录,避免目的基因之外的其他下游序列得到转录和翻译。
67.对于spcas9蛋白的密码子,本技术对其密码子进行了优化,使之完全适应本技术所选用的大肠杆菌高效表达菌株e.coli bl21(de3)的密码子偏好,从而提高cas9蛋白的表达水平。
68.t7启动子如seq id no:1中第5121-5139位核苷酸所示。
69.lac操纵子如seq id no:1中第5140-5164位核苷酸所示。
70.核糖体结合位点如seq id no:1中第5178-5201位核苷酸所示。
71.碱性磷酸酶信号肽的编码序列如seq id no:1中第5209-5271位核苷酸所示。
72.trxa蛋白的编码序列如seq id no:1中第5272-5598位核苷酸所示。
73.his-tag的编码序列如seq id no:1中第5620-5637位核苷酸所示。
74.肠激酶酶切位点的编码序列如seq id no:1中第5638-5652位核苷酸所示。
75.核定位信号的编码序列如seq id no:1中第5656-5670位核苷酸所示。
76.spcas9蛋白的编码序列如seq id no:1中第5701-9801位核苷酸所示。
77.核定位信号的编码序列如seq id no:1中第9802-9849位核苷酸所示。
78.t7终止子如seq id no:1中第9902-9949位核苷酸。
79.具体的,所述特异质粒为质粒pkg-ge4。
80.质粒pkg-ge4中具有seq id no:1中第5121-9949位核苷酸所示的dna分子。
81.具体的,以上任一所述质粒pkg-ge4如seq id no:1所示。
82.本发明还保护以上任一所述方法制备得到的重组猪细胞。
83.所述重组猪细胞为tnip1基因发生突变的重组猪细胞。
84.所述重组猪细胞具体可为表1中基因型为杂合型、双等位基因相同突变型或双等位基因不同突变型的单细胞克隆。
85.本发明还保护所述重组猪细胞在制备银屑病模型猪中的应用。
86.将所述重组猪细胞作为核移植供体细胞进行体细胞克隆,可以得到克隆猪,即为银屑病模型猪。
87.本发明还保护利用所述重组猪细胞制备的模型猪的猪组织,即银屑病组织模型。
88.本发明还保护利用所述重组猪细胞制备的模型猪的猪器官,即银屑病器官模型。
89.本发明还保护利用所述重组猪细胞制备的模型猪的猪细胞,即银屑病细胞模型。
90.本发明还保护所述重组猪细胞、所述银屑病组织模型、所述银屑病器官模型、所述银屑病细胞模型或者所述银屑病模型猪的应用,为如下(d1)或(d2)或(d3)或(d4):
91.(d1)筛选治疗银屑病的药物;
92.(d2)进行银屑病药物的药效评价;
93.(d3)进行银屑病的基因治疗和/或细胞治疗的疗效评价;
94.(d4)研究银屑病的发病机制。
95.以上任一所述猪具体可为从江香猪。
96.以上任一所述猪具体可为初生从江香猪。
97.以上任一所述猪具体可为巴马香猪。
98.以上任一所述猪具体可为初生巴马香猪。
99.以上任一所述银屑病是由tnip1基因突变引起的。
100.猪tnip1基因信息:编码tnfaip3相互作用蛋白1;位于16号染色体;gene id为100271903,sus scrofa。
101.猪tnip1基因编码的蛋白质的氨基酸序列如seq id no:8所示。
102.猪tnip1基因具有seq id no:9所示的dna区段。
103.以上任一所述突变为一个或多个核苷酸的缺失和/或插入和/或替换。
104.以上任一所述突变为一个或多个核苷酸的缺失。
105.以上任一所述突变为一个或多个核苷酸的插入。
106.以上任一所述突变为一个或多个核苷酸的缺失和插入。
107.与现有技术相比,本发明至少具有如下有益效果:
108.(1)本发明研究对象(猪)比其他动物(大小鼠、灵长类)具有更好的应用性。
109.大小鼠等啮齿类动物不论从体型、器官大小、生理、病理等方面都与人相差巨大,无法真实地模拟人类正常的生理、病理状态。研究表明,95%以上在大小鼠中验证有效的药物在人类临床试验中是无效的。就大动物而言,灵长类是与人亲缘关系最近的动物,但其体型小、性成熟晚(6-7岁开始交配),且为单胎动物,群体扩繁速度极慢,饲养成本很高。另外,灵长类动物克隆效率低、难度大、成本高。
110.而猪作为模型动物就没有上述缺点,猪是除灵长类外与人亲缘关系最近的动物,其体型、体重、器官大小等与人相近,在解剖学、生理学、免疫学、营养代谢、疾病发病机制等方面与人类极为相似。同时,猪的性成熟早(4-6个月),繁殖力高,一胎多仔,在2-3年内即可形成一个较大群体。另外,猪的克隆技术非常成熟,克隆及饲养成本也较灵长类低得多。因此猪是非常适合作为人类疾病模型的动物。
111.(2)本发明所构建的载体,使用了能够高效表达目的蛋白的强启动子t7-lac来进行目的蛋白的表达,用细菌周质蛋白碱性磷酸酶(phoa)的信号肽来引导目的蛋白分泌表达至细菌周质腔中,从而与细菌胞内蛋白分离,且分泌到细菌周质腔中的目的蛋白为可溶性表达。同时还采用硫氧还原蛋白trxa与cas9蛋白融合表达,trxa能帮助所共表达的目的蛋白形成二硫键,提高蛋白的稳定性、折叠的正确性,增加目的蛋白的溶解性及活性。为了方便目的蛋白的纯化,设计了his标签,可以通过一步法ni柱亲和层析纯化目的蛋白,极大地简化了目的蛋白的纯化流程。同时在his标签后设计了一个肠激酶酶切位点,便于切除所融合的trxa-his多肽片段,得到天然形式的cas9蛋白。利用带his标签的肠激酶酶切融合蛋白后,可通过一次亲和层析除去trxa-his多肽片段及带his标签的肠激酶,得到天然形式的cas9蛋白,避免了多次纯化透析对目的蛋白的伤害和损耗。同时,本发明也在cas9的n端及c端分别设计了一个nls位点,使cas9能更有效地进入细胞核进行基因编辑。另外,本发明选择了e.coli bl21(de3)菌株为目的蛋白表达菌株,该菌株可高效表达克隆于含有噬菌体t7启动子的表达载体(如pet-32a)的外源基因。同时,对于cas9蛋白的密码子,本发明进行了密码子优化,使之完全适应表达菌株的密码子偏好,从而提高目的蛋白的表达水平。另外,本发明在细菌生长至一定数量后,再用iptg在低温下诱导目的蛋白的表达,可避免目的蛋白过早表达对宿主菌生长的影响,低温下诱导表达也显著提高所表达的目的蛋白的可溶性。经过上述各项优化设计及实验实施,所得到的cas9蛋白活性比商品cas9蛋白有了极显著的提高。
112.(3)采用本发明构建并表达的cas9高效蛋白联合体外转录的grna进行基因编辑,并对cas9和grna的最佳用量配比进行了优化,最终获得基因编辑单细胞克隆的比率高达86.2%,远高于常规的基因编辑效率(10-30%)。
113.(4)利用本发明所得到的靶基因敲除单细胞克隆株进行体细胞核移植动物克隆可直接得到靶基因敲除的克隆猪,并且该基因变异可稳定遗传。
114.在小鼠模型制作中采用的受精卵显微注射基因编辑材料后再进行胚胎移植的方法,因其直接获得基因突变后代的概率比较低,需要进行后代的杂交选育,不太适用于妊娠期较长的大动物(如猪)模型制作。因此,本发明采用技术难度大、挑战性高的原代细胞体外编辑以及cas9蛋白和双grna切割并筛选阳性编辑单细胞克隆的方法,后期再通过体细胞核移植动物克隆技术直接获得相应疾病模型猪,可大大缩短模型猪制作周期并节省人力、物力、财力。
115.本发明采用crispr/cas9技术联合双grna编辑进行了tnip1基因的敲除,模拟银屑病的遗传特征,并获得了tnip1基因敲除的单细胞克隆,为后期通过体细胞核移植动物克隆技术培育银屑病模型猪奠定了基础。本发明将有助于研究并揭示由tnip1基因功能异常所导致的银屑病的发病机制,也可用于进行药物筛选、药效评价、基因治疗及细胞治疗等研究,能够为进一步的临床应用提供有效的实验数据,进而为成功治疗人类银屑病提供有力
的实验手段。本发明对于银屑病药物的研发及揭示该病的发病机制具有重大应用价值。
附图说明
116.图1为质粒pet-32a的结构示意图。
117.图2为质粒pkg-ge4的结构示意图。
118.图3为实施例2中grna与ncn蛋白用量配比优化的电泳图。
119.图4为实施例2中ncn蛋白与商品cas9蛋白的基因编辑效率比较的电泳图。
120.图5为实施例3中用命名为bx4的猪的耳组织提取基因组作为模板采用不同引物对进行pcr扩增的电泳图。
121.图6为实施例3中分别以10只猪的基因组dna为模板采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对进行pcr扩增的电泳图。
122.图7为编号为4的单细胞克隆的正向测序与野生型序列的比对结果。
123.图8为编号为2的单细胞克隆的正向测序与野生型序列的比对结果。
124.图9为编号为15的单细胞克隆的正向测序与野生型序列的比对结果。
125.图10为编号为8的单细胞克隆的正向测序与野生型序列的比对结果。
具体实施方式
126.下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
127.下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。实施例中构建的重组质粒,均已进行测序验证。商品cas9-a蛋白为市售的效果好的cas9蛋白。商品cas9-b蛋白为市售的效果好的cas9蛋白。完全培养液(%为体积比):15%胎牛血清(gibco)+83%dmem培养基(gibco)+1%penicillin-streptomycin(gibco)+1%hepes(solarbio)。细胞培养条件:37℃,5%co2、5%o2的恒温培养箱。
128.实施例2中采用的猪原代成纤维细胞是用初生从江香猪耳组织制备得到的。实施例3和实施例4中采用的猪原代成纤维细胞均是用初生巴马香猪耳组织制备得到的。用猪耳组织制备猪原代成纤维细胞的方法:
①
取猪耳组织0.5g,去除毛发及骨组织,然后用75%酒精浸泡30-40s,然后用含5%(体积比)penicillin-streptomycin(gibco)的pbs缓冲液洗涤5次,然后用pbs缓冲液洗涤一次;
②
用剪刀将组织剪碎,采用5ml 0.1%胶原酶溶液(sigma),37℃消化1h,然后500g离心5min,弃上清;
③
将沉淀用1ml完全培养液重悬,然后铺入含10ml完全培养液并已用0.2%明胶(vwr)封盘的直径为10cm的细胞培养皿中,培养至细胞长满皿底60%左右;
④
完成步骤
③
后,采用胰蛋白酶消化并收集细胞,然后重悬于完全培养液。用于进行后续电转实验。
129.实施例1、ncn蛋白的制备和纯化
130.一、原核cas9高效表达载体的构建
131.质粒pet-32a的结构示意图见图1。
132.质粒pkg-ge4是以质粒pet-32a为出发质粒进行改造得到的。质粒pet32a-t7lac-phoa:sp-trxa-his-ek-nls-spcas9-nls-t7ter(简称质粒pkg-ge4),如seq id no:1所示,为环形质粒,结构示意图见图2。
133.seq id no:1中,第5121-5139位核苷酸组成t7启动子,第5140-5164位核苷酸编码lac操纵子(lac operator),第5178-5201位核苷酸组成核糖体结合位点(rbs),第5209-5271位核苷酸编码碱性磷酸酶信号肽(phoa signal peptide),第5272-5598位核苷酸编码trxa蛋白,第5620-5637位核苷酸编码his-tag(又称为his6标签),第5638-5652位核苷酸编码肠激酶酶切位点(ek酶切位点),第5656-5670位核苷酸编码核定位信号,第5701-9801位核苷酸编码spcas9蛋白,第9802-9849位核苷酸编码核定位信号,第9902-9949位核苷酸组成t7终止子。编码spcas9蛋白的核苷酸已进行针对大肠杆菌bl21(de3)菌株的密码子优化。
134.质粒pkg-ge4的主要改造如下:
①
保留了trxa蛋白的编码区域,trxa蛋白可以帮助所表达的目的蛋白形成二硫键、增加目的蛋白的溶解性及活性;在trxa蛋白的编码区域之前加入碱性磷酸酶信号肽的编码序列,碱性磷酸酶信号肽可以引导所表达的目的蛋白分泌至细菌的膜周质腔中并可被原核周质信号肽酶酶切;
②
在trxa蛋白的编码序列之后增加his-tag的编码序列,his-tag可用于所表达的目的蛋白的富集;
③
在his-tag的编码序列下游增加肠激酶酶切位点ddddk(asp-asp-asp-asp-lys)的编码序列,纯化出的蛋白将在肠激酶作用下去除his-tag和上游所融合的trxa蛋白;
④
插入密码子优化后的适宜大肠杆菌bl21(de3)菌株表达的cas9基因,同时在该基因的上游和下游均增加核定位信号编码序列,增加后期纯化出的cas9蛋白的核定位能力。
135.质粒pkg-ge4中的融合基因如seq id no:1中第5209-9852位核苷酸所示,编码seq id no:2所示的融合蛋白(融合蛋白trxa-his-ek-nls-spcas9-nls,简称为proncn蛋白)。由于碱性磷酸酶信号肽以及肠激酶酶切位点的存在,融合蛋白被肠激酶酶切后形成seq id no:3所示的蛋白质,将seq id no:3所示的蛋白质命名为ncn蛋白。
136.二、诱导表达
137.1、将质粒pkg-ge4导入大肠杆菌bl21(de3),得到重组菌。
138.2、将步骤1得到的重组菌接种至含100μg/ml氨苄青霉素的液体lb培养基,37℃、200rpm振荡培养过夜。
139.3、将步骤2得到的菌液接种至液体lb培养基,30℃、230rpm振荡培养至od
600nm
值=1.0,然后加入异丙基硫代半乳糖苷(iptg)并使其在体系中的浓度为0.5mm,然后25℃、230rpm振荡培养12小时,然后4℃、10000g离心15分钟,收集菌体。
140.4、取步骤3得到的菌体,用pbs缓冲液洗涤。
141.三、融合蛋白trxa-his-ek-nls-spcas9-nls的纯化
142.1、取步骤二得到的菌体,加入粗提缓冲液并悬浮菌体,然后采用均质机进行菌体破碎(1000par循环三次),然后4℃、15000g离心30min,收集上清液,上清液采用0.22μm孔径滤膜过滤,收集滤液。本步骤中,每g湿重的菌体配比10ml粗提缓冲液。
143.粗提缓冲液:含20mm tris-hcl(ph8.0)、0.5m nacl、5mm imidazole、1mm pmsf,余量为ddh2o。
144.2、采用亲和层析纯化融合蛋白。
145.首先采用5个柱体积的平衡液平衡ni-nta琼脂糖柱(流速为1ml/min);然后上样
50ml步骤1得到的滤液(流速为0.5-1ml/min);然后用5个柱体积的平衡液洗涤柱子(流速为1ml/min);然后用5个柱体积的缓冲液洗涤柱子(流速为1ml/min),以去除杂蛋白;然后用10个柱体积的洗脱液以0.5-1ml/min的流速洗脱,收集过柱后溶液(90-100ml)。
146.ni-nta琼脂糖柱:金斯瑞,l00250/l00250-c,填料为10ml。
147.平衡液:含20mm tris-hcl(ph 8.0)、0.5m nacl、5mm imidazole,余量为ddh2o。
148.缓冲液:含20mm tris-hcl(ph 8.0)、0.5m nacl、50mm imidazole,余量为ddh2o。
149.洗脱液:含20mm tris-hcl(ph 8.0)、0.5m nacl、500mm imidazole,余量为ddh2o。
150.四、融合蛋白trxa-his-ek-nls-spcas9-nls的酶切与ncn蛋白的纯化
151.1、取15ml步骤三收集的过柱后溶液,使用amicon超滤管(sigma,ufc9100,容量为15ml)将其浓缩至200μl,然后用25mm tris-hcl(ph8.0)稀释至1ml。采用6个超滤管,共得到6ml。
152.2、将商品来源的具有his6标签的重组牛肠激酶(生工生物,c620031,重组牛肠激酶轻链,带his6标签,recombinant bovine enterokinase light chain,his)加入到步骤1得到的溶液(约6ml)中,25℃酶切16小时。每50μg蛋白量配比加入2个单位的肠激酶。
153.3、取完成步骤2的溶液(约6ml),与480μl ni-nta树脂(金斯瑞,l00250/l00250-c)混匀,在室温下旋转混匀15min,然后7000g离心3min,收集上清液(4-5.5ml)。
154.4、取步骤3得到的上清液,使用amicon超滤管(sigma,ufc9100,容量为15ml)将其浓缩至200μl,然后加入酶贮存液中,调整蛋白浓度为5mg/ml,即为ncn蛋白溶液。
155.经测序,ncn蛋白溶液中的蛋白质,n端15个氨基酸残基如seq id no:3第1至15位所示,即ncn蛋白。
156.用于后续实施例的ncn蛋白由ncn蛋白溶液提供。
157.酶贮存液(ph7.4):含10mm tris,300mm nacl,0.1mm edta,1mm dtt,50%(体积比)甘油,余量为ddh2o。
158.实施例2、ncn蛋白的性能
159.选择靶向ttn基因的2个grna靶点如下:
160.ttn-grna1靶点:agagcacagtcagcctggcg;
161.ttn-grna2靶点:cttccagaattggatctccg。
162.用于鉴定包含ttn基因中grna的靶点片段的引物如下:
163.ttn-f55:tacggaattggggagccagcgga;
164.ttn-r560:caaagttaactctctgtgtct。
165.一、制备grna
166.1、制备ttn-t7-grna1转录模板和ttn-t7-grna2转录模板
167.ttn-t7-grna1转录模板为双链dna分子,如seq id no:4所示。
168.ttn-t7-grna2转录模板为双链dna分子,如seq id no:5所示。
169.2、体外转录得到grna
170.取ttn-t7-grna1转录模板,采用transcript aid t7 high yield transcription kit(fermentas,k0441)进行体外转录,然后用mega clear
tm transcription clean-up kit(thermo,am1908)进行回收纯化,得到ttn-grna1。ttn-grna1为单链rna,如seq id no:6所示。
171.取ttn-t7-grna2转录模板,采用transcript aid t7 high yield transcription kit(fermentas,k0441)进行体外转录,然后用mega clear
tm transcription clean-up kit(thermo,am1908)进行回收纯化,得到ttn-grna2。ttn-grna2为单链rna,如seq id no:7所示。
172.二、grna与ncn蛋白用量配比优化
173.1、共转染猪原代成纤维细胞
174.第一组:将ttn-grna1、ttn-grna2和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:0.5μg ttn-grna1:0.5μg ttn-grna2:4μg ncn蛋白。
175.第二组:将ttn-grna1、ttn-grna2和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:0.75μg ttn-grna1:0.75μg ttn-grna2:4μg ncn蛋白。
176.第三组:将ttn-grna1、ttn-grna2和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2:4μg ncn蛋白。
177.第四组:将ttn-grna1、ttn-grna2和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1.25μg ttn-grna1:1.25μg ttn-grna2:4μg ncn蛋白。
178.第五组:将ttn-grna1和ttn-grna2共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2。
179.共转染采用电击转染的方式,采用哺乳动物核转染试剂盒(neon kit,thermofisher)与neon tm transfection system电转仪(参数设置为:1450v、10ms、3pulse)。
180.2、完成步骤1后,采用完全培养液培养12-18小时,然后更换新的完全培养液进行培养。电转后培养总时间为48小时。
181.3、完成步骤2后,采用胰蛋白酶消化并收集细胞,提取基因组dna,采用ttn-f55和ttn-r560组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳。
182.电泳图见图3。505bp条带为野生型条带(wt),254bp左右(野生型条带505bp理论缺失251bp)为缺失突变条带(mt)。
183.基因缺失突变效率=(mt灰度/mt条带bp数)/(wt灰度/wt条带bp数+mt灰度/mt条带bp数)
×
100%。第一组基因缺失突变效率为19.9%,第二组基因缺失突变效率为39.9%,第三组基因缺失突变效率为79.9%,第四组基因缺失突变效率为44.3%。第五组未发生突变。
184.结果表明,当两个grna与ncn蛋白的质量配比为1:1:4,实际用量为1μg:1μg:4μg时基因编辑效率最高。因此,确定两个grna与ncn蛋白的最适用量为1μg:1μg:4μg。
185.三、ncn蛋白与商品cas9蛋白的基因编辑效率比较
186.1、共转染猪原代成纤维细胞
187.cas9-a组:将ttn-grna1、ttn-grna2和商品cas9-a蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2:4μg cas9-a蛋白。
188.pkg-ge4组:将ttn-grna1、ttn-grna2和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2:4μg ncn蛋白。
189.cas9-b组:将ttn-grna1、ttn-grna2和商品cas9-b蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2:4μg cas9-b蛋白。
190.control组:将ttn-grna1、ttn-grna2共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-grna1:1μg ttn-grna2。
191.共转染采用电击转染的方式,采用哺乳动物核转染试剂盒(neon kit,thermofisher)与neon tm transfection system电转仪(参数设置为:1450v、10ms、3pulse)。
192.2、完成步骤1后,采用完全培养液培养12-18小时,然后更换新的完全培养液进行培养。电转后培养总时间为48小时。
193.3、完成步骤2后,采用胰蛋白酶消化并收集细胞,提取基因组dna,采用ttn-f55和ttn-r560组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳。
194.电泳图见图4。采用商品cas9-a蛋白的基因缺失突变效率为28.5%,采用ncn蛋白的基因缺失突变效率为85.6%,采用商品cas9-b蛋白的基因缺失突变效率为16.6%。
195.结果表明,与采用商品的cas9蛋白相比,采用本发明制备的ncn蛋白使得基因编辑效率显著提高。
196.实施例3、tnip1基因高效grna靶点的筛选
197.猪tnip1基因信息:编码tnfaip3相互作用蛋白1;位于16号染色体;gene id为100271903,sus scrofa。猪tnip1基因编码的蛋白质的氨基酸序列如seq id no:8所示。猪基因组dna中,tnip1基因共有22个外显子,第1编码外显子及其上下游各300bp如seq id no:9所示。
198.质粒pkg-ge3,为环形质粒,如专利申请202010084343.6中的seq id no:2所示。专利申请202010084343.6中的seq id no:2中,第395-680位核苷酸组成cmv增强子,第682-890位核苷酸组成ef1a启动子,第986-1006位核苷酸编码核定位信号(nls),第1016-1036位核苷酸编码核定位信号(nls),第1037-5161位核苷酸编码cas9蛋白,第5162-5209位核苷酸编码核定位信号(nls),第5219-5266位核苷酸编码核定位信号(nls),第5276-5332位核苷酸编码多肽p2a(多肽p2a的氨基酸序列为“atnfsllkqagdveenpgp”,断裂位置为c端开始第一个氨基酸残基和第二个氨基酸残基之间),第5333-6046位核苷酸编码egfp蛋白,第6056-6109位核苷酸编码多肽t2a(多肽t2a的氨基酸序列为“egrgslltcgdveenpgp”,断裂位置为c端开始第一个氨基酸残基和第二个氨基酸残基之间),第6110-6703位核苷酸编码puromycin蛋白(简称puro蛋白),第6722-7310位核苷酸组成wpre序列元件,第7382-7615位核苷酸组成3’ltr序列元件,第7647-7871位核苷酸组成bgh poly(a)signal序列元件。专利申请202010084343.6中的seq id no:2中,第911-6706位核苷酸形成融合基因,表达融合蛋白。由于自剪切多肽p2a和自裂解多肽t2a的存在,融合蛋白自发形成如下三个蛋白:具有cas9蛋白的蛋白、具有egfp蛋白的蛋白和具有puro蛋白的蛋白。
199.pkg-u6grna载体即质粒pkg-u6grna,为环形质粒,如专利申请202010084343.6中的seq id no:3所示。专利申请202010084343.6中的seq id no:3中,第2280-2539位核苷酸组成hu6启动子,第2558-2637位核苷酸用于转录形成grna骨架。使用时,将20bp左右的dna分子(用于转录形成grna的靶序列结合区)插入质粒pkg-u6grna,形成重组质粒,在细胞中重组质粒转录得到grna。
200.一、tnip1基因预设缺失区域及邻近基因组序列保守性分析
201.10只初生巴马香猪,其中雌性6只(分别命名为bc1、bc2、bc3、bc4、bc5、bc6)、雄性4
只(分别命名为bx1、bx2、bx3、bx4)。
202.tnip1-e1-jdf208:actaggatgcctcagtggct;
203.tnip1-e1-jdr601:tcacgattctgggatgctgg;
204.tnip1-e1-jdf235:cccgctgacatactgctgtt;
205.tnip1-e1-jdr713:cttctggaagacggcccatt。
206.用命名为bx4的猪的耳组织提取基因组作为模板,采用不同引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳。电泳图见图5。图5中:组1:采用tnip1-e1-jdf208和tnip1-e1-jdr601组成的引物对;组2:采用tnip1-e1-jdf208和tnip1-e1-jdr713组成的引物对;组3:采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对;组4:采用tnip1-e1-jdf235和tnip1-e1-jdr713组成的引物对。结果表明,优选采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对进行目的片段扩增。
207.分别以10只猪的基因组dna为模板,采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳。电泳图见图6。回收pcr扩增产物并进行测序,将测序结果与公共数据库中的tnip1基因序列进行比对分析。选择10只猪中共有的保守区进行grna靶点的设计。
208.二、筛选靶点
209.通过筛选ngg(避开可能的突变位点)初步筛选到若干靶点,经过预实验进一步从中筛选到4个靶点。
210.4个靶点分别如下:
211.tnip1-e1-grna1靶点:taccggatctacgaccccgg;
212.tnip1-e1-grna2靶点:ataccggatctacgaccccg;
213.tnip1-e1-grna3靶点:cgactgaaggaaaagatgca;
214.tnip1-e1-grna4靶点:gactgaaggaaaagatgcaa。
215.三、制备grna
216.取质粒pkg-u6grna,用限制性内切酶bbsi进行酶切,回收载体骨架(约3kb的线性大片段)。
217.分别合成tnip1-e1-grna1-s和tnip1-e1-grna1-a,然后混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(tnip1-e1-grna1)。质粒pkg-u6grna(tnip1-e1-grna1)表达seq id no:10所示的sgrna
tnip1-e1-grna1
。
218.sgrna
tnip1-e1-grna1
(seq id no:10):
219.uaccggaucuacgaccccggguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuu
220.分别合成tnip1-e1-grna2-s和tnip1-e1-grna2-a,然后混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(tnip1-e1-grna2)。质粒pkg-u6grna(tnip1-e1-grna2)表达seq id no:11所示的sgrna
tnip1-e1-grna2
。
221.sgrna
tnip1-e1-grna2
(seq id no:11):
222.auaccggaucuacgaccccgguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuauca
acuugaaaaaguggcaccgagucggugcuuuu
223.分别合成tnip1-e1-grna3-s和tnip1-e1-grna3-a,然后混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(tnip1-e1-grna3)。质粒pkg-u6grna(tnip1-e1-grna3)表达seq id no:12所示的sgrna
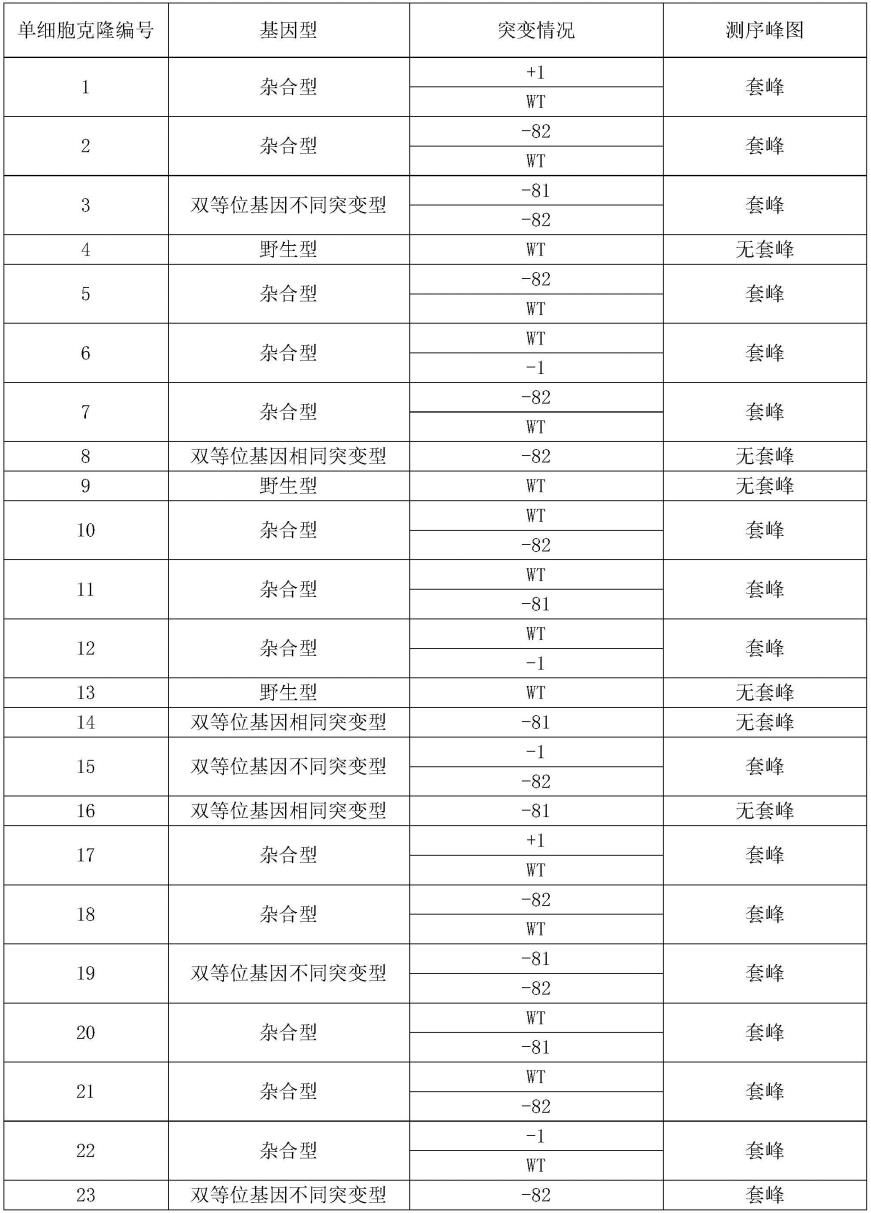
tnip1-e1-grna3
。
224.sgrna
tnip1-e1-grna3
(seq id no:12):
225.cgacugaaggaaaagaugcaguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuu
226.分别合成tnip1-e1-grna4-s和tnip1-e1-grna4-a,然后混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(tnip1-e1-grna4)。质粒pkg-u6grna(tnip1-e1-grna4)表达seq id no:13所示的sgrna
tnip1-e1-grna4
。
227.sgrna
tnip1-e1-grna4
(seq id no:13):
228.gacugaaggaaaagaugcaaguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuu
229.tnip1-e1-grna1-s:caccgtaccggatctacgaccccgg;
230.tnip1-e1-grna1-a:aaacccggggtcgtagatccggtac;
231.tnip1-e1-grna2-s:caccgataccggatctacgaccccg;
232.tnip1-e1-grna2-a:aaaccggggtcgtagatccggtatc;
233.tnip1-e1-grna3-s:caccgcgactgaaggaaaagatgca;
234.tnip1-e1-grna3-a:aaactgcatcttttccttcagtcgc;
235.tnip1-e1-grna4-s:caccgactgaaggaaaagatgcaa;
236.tnip1-e1-grna4-a:aaacttgcatcttttccttcagtc。
237.tnip1-e1-grna1-s、tnip1-e1-grna1-a、tnip1-e1-grna2-s、tnip1-e1-grna2-a、tnip1-e1-grna3-s、tnip1-e1-grna3-a、tnip1-e1-grna4-s、tnip1-e1-grna4-a均为单链dna分子。
238.四、不同靶点组合的编辑效率比较
239.1、共转染
240.第一组:将质粒pkg-u6grna(tnip1-e1-grna1)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(tnip1-e1-grna1):1.08μg质粒pkg-ge3。
241.第二组:将质粒pkg-u6grna(tnip1-e1-grna2)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(tnip1-e1-grna2):1.08μg质粒pkg-ge3。
242.第三组:将质粒pkg-u6grna(tnip1-e1-grna3)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(tnip1-e1-grna3):1.08μg质粒pkg-ge3。
243.第四组:将质粒pkg-u6grna(tnip1-e1-grna4)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(tnip1-e1-grna4):1.08μ
g质粒pkg-ge3。
244.第五组:猪原代成纤维细胞,同等电转参数不加质粒进行电转操作。
245.共转染采用电击转染的方式,采用哺乳动物核转染试剂盒(neon kit,thermofisher)与neon tm transfection system电转仪(参数设置为:1450v、10ms、3pulse)。
246.2、完成步骤1后,采用完全培养液培养12-18小时,然后更换新的完全培养液进行培养。电转后培养总时间为48小时。
247.3、完成步骤2后,采用胰蛋白酶消化并收集细胞,裂解细胞,提取基因组dna,采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳。检测细胞靶基因突变情况。
248.将目的产物切胶回收后送测序公司进行测序,然后将测序结果利用网页版synthego ice工具分析测序峰图得出不同靶点的基因编辑效率。第一组、第二组、第三组、第四组的基因编辑效率依次为8%、34%、38%、32%,第五组未发生基因编辑。结果表明,tnip1-e1-grna2靶点和tnip1-e1-grna3靶点编辑效率较高。
249.实施例4、制备tnip1基因敲除的巴马香猪单细胞克隆
250.选用实施例3中筛到的两个高效grna靶点(tnip1-e1-grna2和tnip1-e1-grna3)。
251.一、制备grna
252.1、制备tnip1-t7-grna2转录模板和tnip1-t7-grna3转录模板
253.tnip1-t7-grna2转录模板为双链dna分子,如seq id no:14所示。
254.tnip1-t7-grna3转录模板为双链dna分子,如seq id no:15所示。
255.2、体外转录得到grna
256.取tnip1-t7-grna2转录模板,采用transcript aid t7 high yield transcription kit(fermentas,k0441)进行体外转录,然后用mega clear
tm transcription clean-up kit(thermo,am1908)进行回收纯化,得到tnip1-grna2。tnip1-grna2为单链rna,如seq id no:16所示。
257.取tnip1-t7-grna3转录模板,采用transcript aid t7 high yield transcription kit(fermentas,k0441)进行体外转录,然后用mega clear
tm transcription clean-up kit(thermo,am1908)进行回收纯化,得到tnip1-grna3。tnip1-grna3为单链rna,如seq id no:17所示。tnip1-grna2(seq id no:16):
258.ggauaccggaucuacgaccccgguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuutnip1-grna3(seq id no:17):
259.ggcgacugaaggaaaagaugcaguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuu
260.二、转染猪原代成纤维细胞
261.1、将tnip1-grna2、tnip1-grna3和ncn蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg tnip1-grna2:1μg tnip1-grna3:4μg ncn蛋白。共转染采用电击转染的方式,采用哺乳动物核转染试剂盒(neon kit,thermofisher)与neon tm transfection system电转仪(参数设置为:1450v、10ms、3pulse)。
262.2、完成步骤1后,采用完全培养液培养16-18小时,然后更换新的完全培养液进行
培养。电转后培养总时间为48小时。
263.3、完成步骤2后,采用胰蛋白酶消化并收集细胞,然后用完全培养液洗涤,然后用完全培养液重悬,然后分别挑取各个单克隆转移到96孔板中(每个孔1个细胞,每个孔中装有100μl完全培养液),培养2周(每2-3天更换新的完全培养液)。
264.4、完成步骤3后,采用胰蛋白酶消化并收集细胞(每孔得到的细胞,约2/3接种到装有完全培养液的6孔板中,剩余的1/3收集在1.5ml离心管中)。
265.5、取步骤4的6孔板,培养直至细胞长至80%汇合度,采用胰蛋白酶消化并收集细胞,使用细胞冻存液(90%完全培养基+10%dmso,体积比)将细胞冻存。
266.6、取步骤4的离心管,取细胞,进行细胞裂解并提取基因组dna,采用tnip1-e1-jdf235和tnip1-e1-jdr601组成的引物对进行pcr扩增,然后进行电泳。将猪原代成纤维细胞作为野生型对照(wt)。
267.7、完成步骤6后,回收pcr扩增产物并测序。
268.猪原代成纤维细胞的测序结果只有一种,其基因型为野生型(也可称为纯合野生型)。如果某一单细胞克隆的测序结果有两种,一种与猪原代成纤维细胞的测序结果一致,另一种与猪原代成纤维细胞的测序结果相比发生了突变(突变包括一个或多个核苷酸的缺失、插入或替换),该单细胞克隆的基因型为杂合型;如果某一单细胞克隆的测序结果为两种,均与猪原代成纤维细胞的测序结果相比发生了突变(突变包括一个或多个核苷酸的缺失、插入或替换),该单细胞克隆的基因型为双等位基因不同突变型;如果某一单细胞克隆的测序结果为一种,且与猪原代成纤维细胞的测序结果相比发生了突变(突变包括一个或多个核苷酸的缺失、插入或替换),该单细胞克隆的基因型为双等位基因相同突变型;如果某一单细胞克隆的测序结果为一种,且与猪原代成纤维细胞的测序结果一致,该单细胞克隆的基因型为野生型(也可称为纯合野生型)。
269.结果见表1。编号为4、9、13、27的单细胞克隆的基因型为野生型。编号为1、2、5、6、7、10、11、12、17、18、20、21、22、26、29的单细胞克隆的基因型为杂合型。编号为3、15、19、23、24、25、28的单细胞克隆的基因型为双等位基因不同突变型。编号为8、14、16的单细胞克隆的基因型为双等位基因相同突变型。得到tnip1基因编辑单细胞克隆的比率为86.2%。
270.示例性的测序比对结果见图7至图10。图7是编号为4的单细胞克隆的正向测序与野生型序列的比对结果,判定为野生型。图8是编号为2的单细胞克隆的正向测序与野生型序列的比对结果,判定为杂合型。图9是编号为15的单细胞克隆的正向测序与野生型序列的比对结果,为双等位基因不同突变型。图10是编号8的单细胞克隆正向测序与野生型序列的比对结果,为双等位基因相同突变型。
271.表1 tnip1基因编辑单细胞克隆的基因型测定结果
272.[0273][0274]
上述杂合型、双等位基因相同突变型及双等位基因不同突变型的单细胞克隆均为目标单细胞克隆。将细胞作为核移植供体细胞进行体细胞克隆,可以得到克隆猪,即为银屑病模型猪。
[0275]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1