一种诺乙雄龙的制备方法与流程
1.本发明涉及药物合成技术领域,尤其涉及一种诺乙雄龙的制备方法。
背景技术:
2.诺乙雄龙(norethandrolone,19-去甲-17α-乙基睾酮)是一种与去甲睾酮(诺龙)结构紧密相关的合成代谢类固醇。这种口服合成代谢类固醇的活性为轻度至中度,并有可分辨的雄激素和雌激素成分。这种类固醇本质上是被改性的(烷基化的),且使口服给药变得可行。诺乙雄龙是在1954年首次出现,它是由searle研发出来的药物,在20世纪50年代后期以nilevar品牌的名义被引入美国的处方药市场。nilevar被列出的适应症包括手术的准备和恢复,严重或长时间的疾病,神经性厌食,严重烧伤和创伤,褥疮溃疡,骨质疏松症,骨折愈合,胃肠道疾病,以及成人和儿童各种形式的营养不良。
3.目前报道的用于制备诺乙雄龙的合成路线,最常见的起始物是甲基双酮,专利(wo 2018185783;wo 2020176843)报道了以甲基双酮为起始物,经过炔化和氢化两步反应制备诺乙雄龙,见式1,该方法的缺点是第一步直接对甲基双酮进行炔化,3位上的酮羰基同样也会被炔化产生杂质,而且氢化时4,5位双键也可以被还原产生杂质,由于这些副反应会导致收率偏低和质量难以控制,不利于工业化大生产。
[0004][0005]
djerassi,c.等(j.a m.chem.soc,steroids.liv.synthesis of 19-nor-17a-ethynyltestosterone and 19-nor-17cr-methyltestosterone,1954,76,4092-4094)和专利(cn 104788524 b)报道了甲基双酮先通过醚化反应保护3位酮羰基,使得炔化时3位酮羰基不被炔化产生杂质,再水解得到炔诺酮,见式2,虽然该方法解决了甲基双酮到炔诺酮的3位炔化杂质的产生,但是仍然没有解决氢化时4,5位双键也可以被还原产生杂质的问题,而且增加了2步反应,因此总收率仍然没有提高(总收率为47%~65%)。
[0006]
技术实现要素:
[0007]
本发明的目的在于提供一种诺乙雄龙的制备方法,能够减少反应杂质并提高反应收率。
[0008]
为了实现上述发明目的,本发明提供以下技术方案:
[0009]
本发明提供了一种诺乙雄龙的制备方法,包括以下步骤:
[0010]
将甲基双酮、原甲酸类酯化合物、对甲苯磺酸和醇混合,进行醚化反应,得到醚化产物;
[0011]
将所述醚化产物、格氏试剂溶液和第一有机溶剂混合,进行格氏加成反应,得到格氏产物;
[0012]
将所述格氏产物、酸和第二有机溶剂混合,进行水解反应,得到诺乙雄龙。
[0013]
优选的,所述原甲酸类酯化合物包括原甲酸三甲酯或原甲酸三乙酯;所述醇包括甲醇或乙醇。
[0014]
优选的,所述甲基双酮和对甲苯磺酸的质量比为1:(0.03~0.06);所述甲基双酮与原甲酸类酯化合物的质量比为1:(5~6);所述甲基双酮与醇的质量比为1:(3~5)。
[0015]
优选的,所述醚化反应的温度为50~60℃,时间为5~6h。
[0016]
优选的,所述格氏试剂溶液中格氏试剂包括乙基溴化镁或乙基氯化镁;所述格氏试剂溶液的浓度为1mol/l;所述第一有机溶剂为四氢呋喃。
[0017]
优选的,所述醚化产物与格氏试剂溶液的质量比为1:(4~5);所述醚化产物与第一有机溶剂的质量比为1:(8~10)。
[0018]
优选的,所述格氏加成反应的温度为0~10℃,时间为3~4h。
[0019]
优选的,所述酸为盐酸;所述盐酸的浓度为6mol/l;所述第二有机溶剂为四氢呋喃。
[0020]
优选的,所述格氏产物与酸的质量比为1:(1~2);所述格氏产物与第二有机溶剂的质量比为1:(8~10)。
[0021]
优选的,所述水解反应的温度为20~30℃,时间为5~6h。
[0022]
本发明提供了一种诺乙雄龙的制备方法,包括以下步骤:将甲基双酮、原甲酸类酯化合物、对甲苯磺酸和醇混合,进行醚化反应,得到醚化产物;将所述醚化产物、格氏试剂溶
液和第一有机溶剂混合,进行格氏加成反应,得到格氏产物;将所述格氏产物、酸和第二有机溶剂混合,进行水解反应,得到诺乙雄龙。
[0023]
本发明以甲基双酮作为起始原料,依次经过3位羰基醚化、17位羰基格式反应和3位烷氧基水解三个步骤制备诺乙雄龙,先通过醚化保护3位羰基,然后在17位羰基通过格式反应直接上烷基,无需氢化过程,有效减少了3位产生的杂质和4,5位双键氢化产生的杂质,提高反应总收率至85~95%。
[0024]
本发明所用试剂便宜易得,成本低,合成路线较短,各步反应操作简便。
[0025]
此外,背景技术中式1和式2的反应路线均使用炔气进行炔化反应,氢气进行氢化反应,使用易燃易爆危险气体,增加了工业化大生产的风险。本发明避免使用炔气和氢气等易燃易爆气体,降低了生产风险,有利于工业化大生产。
[0026]
进一步的,本发明以四氢呋喃作为溶剂,比乙醚的安全性高,且在0~10℃低温条件进行格氏加成反应,副反应少,因此相对于乙醚回流条件,减少了杂质的产生。
附图说明
[0027]
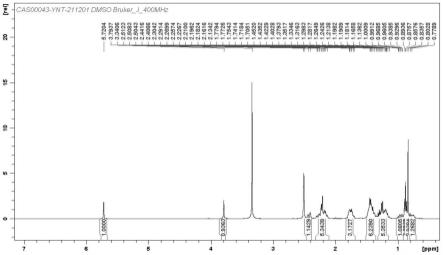
图1为实施例1制备的诺乙雄龙的氢谱图;
[0028]
图2为实施例1制备的诺乙雄龙的氢谱局部放大图;
[0029]
图3为实施例1制备的诺乙雄龙的质谱图;
[0030]
图4为实施例1制备的诺乙雄龙的hplc谱图。
具体实施方式
[0031]
本发明提供了一种诺乙雄龙的制备方法,包括以下步骤:
[0032]
将甲基双酮、原甲酸类酯化合物、对甲苯磺酸和醇混合,进行醚化反应,得到醚化产物;
[0033]
将所述醚化产物、格氏试剂溶液和第一有机溶剂混合,进行格氏加成反应,得到格氏产物;
[0034]
将所述格氏产物、酸和第二有机溶剂混合,进行水解反应,得到诺乙雄龙。
[0035]
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
[0036]
本发明将甲基双酮、原甲酸类酯化合物、对甲苯磺酸和醇混合,进行醚化反应,得到醚化产物。
[0037]
在本发明中,所述原甲酸类酯化合物优选包括原甲酸三甲酯或原甲酸三乙酯;所述醇优选包括甲醇或乙醇;所述甲基双酮和对甲苯磺酸的质量比优选为1:(0.03~0.06),更优选为1:0.045;所述甲基双酮与原甲酸类酯化合物的质量比优选为1:(5~6),更优选为1:5.5;所述甲基双酮与醇的质量比优选为1:(3~5),更优选为1:4。本发明利用原甲酸类酯化合物除去反应体系中产生的水分;本发明利用对甲苯磺酸作为催化剂。
[0038]
在本发明中,所述甲基双酮(记为化合物1)、原甲酸类酯化合物、对甲苯磺酸和醇混合的过程优选为将原甲酸类酯化合物、醇和甲基双酮室温搅拌后,加入对甲苯磺酸。
[0039]
在本发明中,所述醚化反应的温度优选为50~60℃,时间优选为5~6h,更优选为5.5h;所述醚化反应过程中,醇与甲基双铜的3位酮羰基发生醚化。
[0040]
本发明优选采用tlc监测反应;所用tlc试剂优选为石油醚和丙酮;所述石油醚和
丙酮的体积比优选为5:1。
[0041]
完成所述醚化反应后,本发明优选将所得产物降温至室温,加入三乙胺调ph值为7~8,过滤,使用乙酸乙酯洗涤滤饼,干燥后,得到醚化产物(记为化合物2);所述干燥的温度优选为50~60℃,更优选为55℃。本发明对所述过滤和洗涤的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0042]
得到醚化产物后,本发明将所述醚化产物、格氏试剂溶液和第一有机溶剂混合,进行格氏加成反应,得到格氏产物。
[0043]
在本发明中,所述格氏试剂溶液中格氏试剂优选包括乙基溴化镁或乙基氯化镁;所述格氏试剂溶液的浓度优选为1mol/l;所述格氏试剂溶液所用溶剂优选为四氢呋喃。
[0044]
在本发明中,所述醚化产物与格氏试剂溶液的质量比优选为1:(4~5),更优选为1:(4.53~4.69);所述第一有机溶剂优选为无水四氢呋喃;所述醚化产物与第一有机溶剂的质量比优选为1:(8~10),更优选为1:(9~9.38)。
[0045]
在本发明中,所述醚化产物、格氏试剂溶液和第一有机溶剂混合的过程优选为:在搅拌条件下向第一有机溶剂中加入醚化产物至完全溶解,通n2置换空气,控制温度为0~10℃,更优选为5℃,滴加格氏试剂溶液,密闭体系,进行格氏加成反应。本发明对所述搅拌和滴加的速率没有特殊的限定,按照本领域熟知的过程进行即可。
[0046]
在本发明中,所述格氏加成反应优选在氮气保护条件下进行;所述格氏加成反应的温度为0~10℃,更优选为5℃,时间为3~4h,更优选为3.5h。在所述格氏加成反应过程中,格氏试剂与醚化产物的17位酮羰基进行格氏加成。
[0047]
本发明优选采用tlc监测反应;所用tlc试剂优选为石油醚和丙酮;所述石油醚和丙酮的体积比优选为4:1。
[0048]
完成所述格氏加成反应后,本发明优选控温0~10℃,更优选为5℃,滴加无水乙醇淬灭反应,加入水,用冰醋酸调ph值为5~6,负压浓缩至剩余物料质量与浓缩前物料的质量比为(0.15~0.25):1,过滤,水洗至中性,干燥后,得到格氏产物(记为化合物3);所述干燥的温度优选为50~60℃,更优选为55℃。本发明对所述淬灭、负压浓缩、过滤和水洗的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0049]
得到格氏产物后,本发明将所述格氏产物、酸和第二有机溶剂混合,进行水解反应,得到诺乙雄龙。
[0050]
在本发明中,所述酸优选为盐酸;所述盐酸的浓度优选为6mol/l;所述第二有机溶剂优选为四氢呋喃。
[0051]
在本发明中,所述格氏产物与酸的质量比优选为1:(1~2),更优选为1:(1.5~1.76);所述格氏产物与第二有机溶剂的质量比优选为1:(8~10),更优选为1:(8.8~9)。
[0052]
本发明对所述格氏产物、酸和第二有机溶剂混合的过程没有特殊的限定,按照本领域熟知的过程将物料混合均匀即可。
[0053]
在本发明中,所述水解反应的温度优选为20~30℃,更优选为25℃,时间优选为5~6h,更优选为5.5h;在所述水解过程中,在盐酸作用下,格氏产物的3位醚键水解形成诺乙雄龙。
[0054]
本发明优选采用tlc监测反应;所用tlc试剂优选为石油醚和丙酮;所述石油醚和丙酮的体积比优选为4:1。
[0055]
完成所述水解反应后,本发明优选采用质量浓度为10%的氢氧化钠水溶液调节ph值至7~8,负压浓缩至剩余物料质量与浓缩前物料的质量比为(0.2~0.3):1,过滤后,水洗滤饼至中性,干燥后,得到诺乙雄龙(记为化合物4);本发明对所述负压浓缩、过滤、水洗和干燥的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0056]
在本发明中,制备诺乙雄龙的反应式见式3:
[0057][0058]
式3。
[0059]
在上述式3中,化合物2中r为醚化反应所用醇所对应的基团种类,本发明中所用醇优选为甲醇或乙醇,对应r为甲基或乙基。
[0060]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0061]
实施例1
[0062]
1)向反应瓶中加入原甲酸三甲酯600g、甲醇500g和甲基双酮(化合物1)100g,室温搅拌后加入对甲苯磺酸6g,升温至60℃,tlc监测反应(石油醚:丙酮=5:1,体积比),反应5h后,降温至室温,加入三乙胺调ph值为7,过滤,加入50g乙酸乙酯洗涤滤饼,在55℃烘干,得到96g化合物2,重量收率96%;
[0063]
2)向反应瓶中加入无水四氢呋喃900g,搅拌条件下加入96g化合物2至完全溶解,通n2置换空气,控制温度在5℃,滴加450g乙基溴化镁四氢呋喃溶液(浓度为1mol/l),密闭体系,5℃保温常压反应,tlc监测反应(石油醚:丙酮=4:1,体积比),3h后反应完毕,控温5℃,滴加180g无水乙醇淬灭反应,加入450g水,用冰醋酸调ph值为6,负压浓缩至剩余物料质量400g,过滤,水洗至中性,55℃烘干,得到92g化合物3,重量收率96%;
[0064]
3)在25℃向反应瓶中加入四氢呋喃810g、92g化合物3和162g盐酸(6mol/l),控温25℃搅拌,tlc监测反应(石油醚:丙酮=4:1,体积比),5h后反应完毕,用质量浓度为10%的氢氧化钠水溶液调ph值至7,负压浓缩至剩余物料质量350g,过滤,水洗滤饼至中性,烘干,得到86.6g诺乙雄龙(化合物4),总收率94%。
[0065]
实施例2
[0066]
1)向反应瓶中加入原甲酸三甲酯500g、甲醇300g和甲基双酮(化合物1)100g室温搅拌后,加入对甲苯磺酸3g,升温至60℃,tlc监测反应(石油醚:丙酮=5:1,体积比),反应6h后,降温至室温,加入三乙胺调ph值为7,过滤,采用50g乙酸乙酯洗涤滤饼,55℃烘干,得100g化合物2,重量收率100%;
[0067]
2)向反应瓶中加入无水四氢呋喃800g,搅拌条件下加入100g化合物2至完全溶解,通n2置换空气,控制温度在5℃,滴加400g乙基溴化镁四氢呋喃溶液(1mol/l),密闭体系,5℃保温常压反应,tlc监测反应(石油醚:丙酮=4:1,体积比),4h后反应完毕,控温5℃,滴加
200g无水乙醇淬灭反应,加入500g水,用冰醋酸调ph值为6,负压浓缩至剩余物料质量400g,过滤,水洗至中性,55℃烘干,得到100g化合物3,重量收率100%;
[0068]
3)在25℃向反应瓶中加入四氢呋喃800g、100g化合物3和100g盐酸(6mol/l),控温25℃搅拌,tlc监测反应(石油醚:丙酮=4:1,体积比),6h后反应完毕,用质量浓度为10%的氢氧化钠水溶液调ph值至7,负压浓缩至剩余物料质量350g,过滤,水洗滤饼至中性,烘干,得到95g诺乙雄龙(化合物4),总收率95%。
[0069]
实施例3
[0070]
1)向反应瓶中加入原甲酸三乙酯550g、乙醇400g和甲基双酮(化合物1)100g,室温搅拌后加入对甲苯磺酸4.5g,升温至50℃,tlc监测反应(石油醚:丙酮=5:1,体积比),反应5.5h后,降温至室温,加入三乙胺调ph值为8,过滤,采用50g乙酸乙酯洗涤滤饼,55℃烘干,得95g化合物2,重量收率95%;
[0071]
2)向反应瓶中加入无水四氢呋喃855g,搅拌条件下加入95g化合物2至完全溶解,通n2置换空气,控制温度在5℃,滴加430g乙基氯化镁四氢呋喃溶液(1mol/l),密闭体系,5℃保温常压反应,tlc监测反应(石油醚:丙酮=4:1,体积比),3.5h后反应完毕,控温5℃,滴加190g无水乙醇淬灭反应,加入500g水,用冰醋酸调ph值为6,负压浓缩至剩余物料质量400g,过滤,水洗至中性,55℃烘干,得到90g化合物3,重量收率95%;
[0072]
3)在25℃向反应瓶中加入四氢呋喃810g、90g化合物3和135g盐酸(6mol/l),控温25℃搅拌,tlc监测反应(石油醚:丙酮=4:1,体积比),5.5h后反应完毕,用质量浓度为10%氢氧化钠水溶液调ph值至7,负压浓缩至剩余物料质量350g,过滤,水洗滤饼至中性,烘干,得到85g诺乙雄龙(化合物4),总收率94%。
[0073]
表征
[0074]
对实施例1制备的诺乙雄龙进行核磁表征,所得谱图见图1~3,所得氢谱和质谱数据:
[0075]1hnmr(400mhz,d6-dmso)δ5.72(s,1h,ch=c),3.79(s,1h,-oh),2.44-2.40(m,1h),2.30-2.13(m,5h),1.80-1.70(m,3h),1.50-1.37(m,6h),1.31-1.16(m,5h),1.00-0.92(m,1h),0.89-0.85(t,j=7.2hz,3h,ch2ch3),0.83(s,3h,ch3),0.80-0.72(m,1h);
[0076]
ms(m/z)calcd forc
20h30
o2(m+h)
+
303,found 303。
[0077]
对实施例1制备的诺乙雄龙进行hplc检测,hplc检测条件:样品浓度1g/ml,溶剂:40wt%乙腈,流动相a:100wt%水,b:100%乙腈,流速:0.5ml/min,检测波长240nm,柱温:30℃,进样量2ul,运行时间:25min,色谱柱:j-01-02inertsil ods-2hp 5um 2.1*150mm(up),等度/梯度:b泵0~10min,42~42%;10~16min,42~90%;16~19min,90~90%;19~20min,90~42%;20~25min,42-42%,所得结果见图4。
[0078]
由图4可知,所制备的诺乙雄龙产物纯度为99.8%。
[0079]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1